Article Text

Abstract
The complexity of the care environment, the emergent nature, and the severity of patient injury make conducting clinical trauma research challenging. These challenges hamper the ability to investigate potentially life-saving research that aims to deliver pharmacotherapeutics, test medical devices, and develop technologies that may improve patient survival and recovery. Regulations intended to protect research subjects impede scientific advancements needed to treat the critically ill and injured and balancing these regulatory priorities is challenging in the acute setting. This scoping review attempted to systematically identify what regulations are challenging in conducting trauma and emergency research. A systematic search of PubMed was performed to identify studies published between 2007 and 2020, from which 289 articles that address regulatory challenges in conducting research in emergency settings were included. Data were extracted and summarized using descriptive statistics and a narrative synthesis of the results. The review is reported in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses extension for Scoping Reviews guidelines. Most articles identified were editorial/commentary (31%) and published in the USA (49%). Regulatory factors addressed in the papers were categorized under 15 regulatory challenge areas: informed consent (78%), research ethics (65%), institutional review board (55%), human subjects protection (54%), enrollment (53%), exception from informed consent (51%), legally authorized representative (50%), patient safety (41%), community consultation (40%), waiver of informed consent (40%), recruitment challenges (39%), patient perception (30%), liability (15%), participant incentives (13%), and common rule (11%). We identified several regulatory barriers to conducting trauma and emergency research. This summary will support the development of best practices for investigators and funding agencies.
- research
- informed consent
- policy
- vulnerable populations
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
The complexity of the care environment, the emergent nature, and the severity of patient injury make conducting clinical trauma research challenging.1 These challenges hamper the ability to conduct potentially life-saving research that aims to deliver pharmacotherapeutics, test medical devices, and develop technologies that may improve patient survival and recovery.2 Regulations intended to protect research subjects impede scientific advancements needed to treat the critically ill and injured and balancing these regulatory priorities is challenging in the acute setting. As a scoping review, this work attempted to systematically identify and summarize regulatory challenges in conducting trauma and emergency research.3 Scoping reviews allow researchers to conduct a comprehensive search to gather information on a specific topic focused area. The information in this review synthesizes the evidence and assesses the size and scope of available research surrounding this topic.
In 2014, the National Trauma Institute, now known as the Coalition for National Trauma Research (CNTR), surveyed 16 federally funded investigators to identify facilitators and barriers to conducting trauma research.4 Forty percent of the investigators reported challenges in obtaining regulatory approval. Several investigators encountered difficulties navigating the requirements for the Department of Defense (DoD) Human Research Protections Office (HRPO) approval processes. Multisite studies were delayed due to multiple institutional review board (IRB) reviews with conflicting revisions. The mean number of days from funding selection to IRB approval was 210 days, while the mean number of days from funding selection to the HRPO approval was 401 days. Other multisite studies have reported timelines of up to a year to obtain approval.5–7 These data are evidence of the challenges investigators encounter while initiating trauma studies and the need for guidance.
In the 2016 National Academies of Sciences, Engineering and Medicine (NASEM) report that called for a national trauma care system, the authors concluded that ‘a learning trauma care system cannot function optimally in the current federal regulatory environment’.2 Identified barriers included ambiguity in the interpretation of federal regulations, regulatory silence on specific issues, a lack of flexibility in interpreting data that may lead to regulatory approval for new therapies, and the various applicable federal regulations. The NASEM report also recommended identifying regulatory barriers to trauma research and suggested that federal agencies work ‘to revise research regulations and reduce misinterpretation of the regulations through policy statements’.2
In 2018, the CNTR received DoD funding to develop the National Trauma Research Action Plan (NTRAP) (Contract No. W81XWH-18-C-0179). The NTRAP builds on the NASEM report with the understanding that reducing regulatory challenges requires a resourced, coordinated, and multidisciplinary approach. NTRAP’s three aims were to: (1) perform a gap analysis of trauma research; (2) define optimal metrics to assess long-term outcomes in injured patients; and (3) identify trauma research regulatory barriers, develop regulatory best practices, and collaborate with federal entities to define optimal end points. On completion, the NTRAP will provide a road map for investigators and funding agencies to prioritize trauma research across the continuum of care. The objective of this study and analysis is to conduct a literature review to identify barriers and misinterpretations regarding the conduct of trauma research in emergency settings. Although researchers may be aware of research barriers, this scoping review details how often these barriers have appeared in the literature since 2007; therefore, providing a strong foundation of themes to prioritize and guide future direction on next steps.
Methods
Protocol
Our scoping review protocol was developed using the Preferred Reporting Items for Systematic Reviews and Meta-Analyses extension for Scoping Reviews (PRISMA-ScR) guidelines and revised by members of the NTRAP Publications Committee for scientific content and consistency of data interpretation with previous NTRAP publications.
Eligibility criteria
Studies were included if they were in English and published from January 1, 2007 through November 4, 2020. Selecting 2007 as the beginning of the published date range aligned with the year that Congress passed the Food and Drug Administration Amendments Act of 2007, requiring more clinical trials registration, sharing additional trial information, and the submission of summary results, including adverse events.8 Regulatory issues that were not applicable in the USA were excluded from the thematic analysis.
Information sources
NTRAP investigators searched articles in PubMed using the search strategy detailed in online supplemental appendix A. Maintained by the National Center for Biotechnology Information, PubMed is the best source to capture the majority of the published work in this area.9 The scoping review included human subjects protection issues in conducting trauma and emergency research using a combination of text words and Medical Subject Headings terms. The search strategy was developed in collaboration with an experienced librarian. Search results were downloaded and exported into EndNote (Thomson Reuters, New York, USA). The electronic database search was supplemented by checking the citation lists of included articles. Covidence, a Cochrane Review production tool, was used for article screening and data extraction due to its ability to manage and streamline the process.10
Supplemental material
Selection of sources of evidence
All citations were imported from Endnote into Covidence, and duplicate records were removed. The selection of sources of evidence was based on the inclusion/exclusion criteria and carried out manually by six reviewers in three stages:
Title and abstract screening (CLV, MAP)
Full-text review (CLV, MAP)
Extraction (ANM, AZ, CS, AT) with oversight and quality checking on all cases (CLV)
Disagreements on study selection were resolved by the consensus of two researchers (CLV, MAP).
Data charting process
A data charting form was developed by two researchers (CLV and MAP) to determine variable extraction. Data specific to the review question and necessary for the narrative synthesis were extracted, including study characteristics, population characteristics, regulatory body discussed, and regulatory challenges addressed. The form was then reviewed by two additional researchers (EMB and JPH-E) for the inclusion of other critical variables (see online supplemental appendix B). These variables were used to create a data dictionary for the extraction phase. A training session was held with the review team, and ongoing training sessions were conducted to ensure that key points of clarification were examined. A team of four research associates (ANM, AZ, CS, AT) extracted the data, discussed the results, and updated the data charting form in an iterative process. One reviewer independently extracted the data from each included article, and a lead researcher (CLV) conducted quality assurance checks for all studies. Quality assurance checks were conducted using the data charting form to confirm that all the information was extracted and that each regulatory challenge discussed in the article was notated.
Data items and synthesis of results
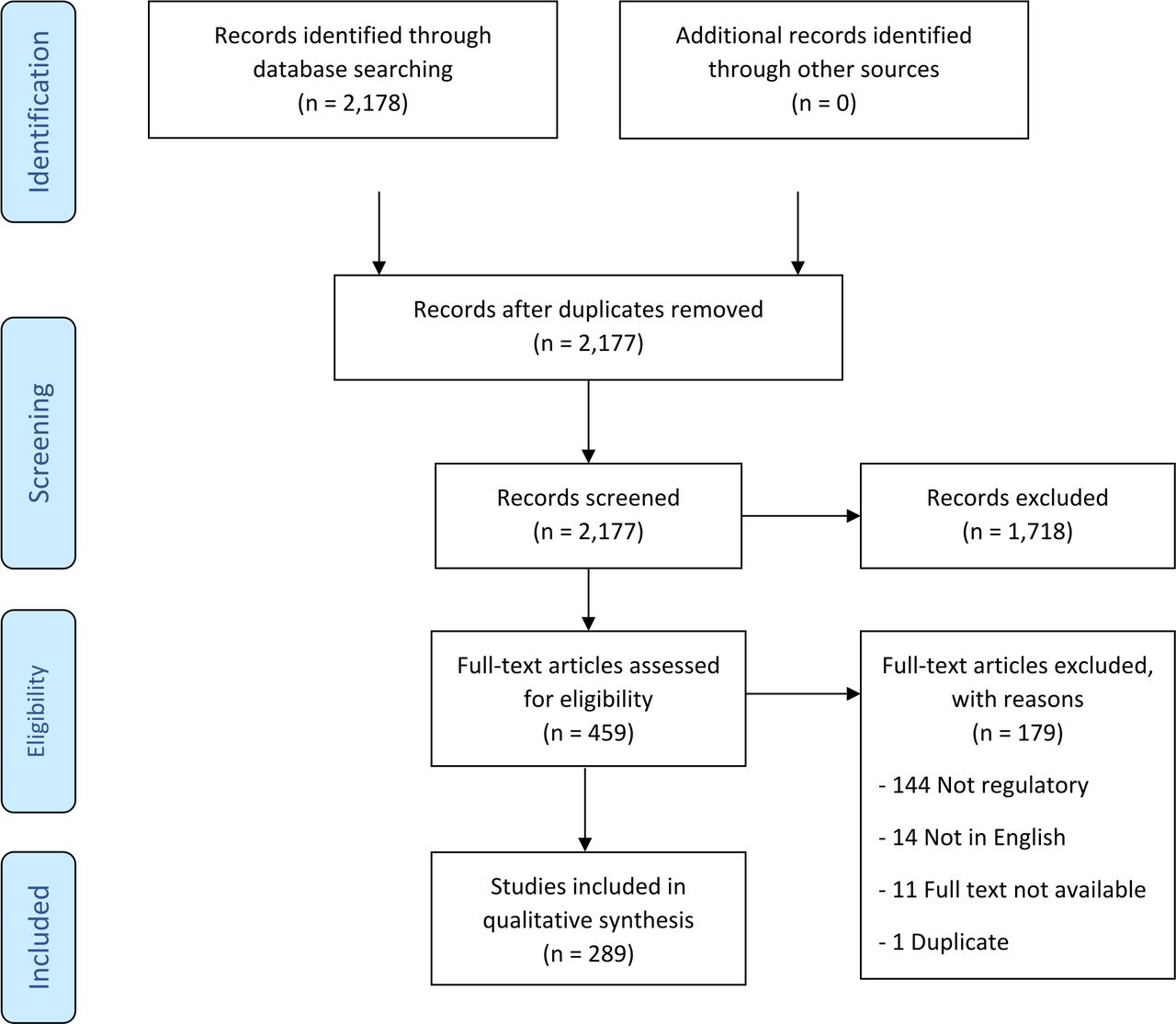
We abstracted data on article characteristics (eg, author, year of publication, country of origin, study type, keywords), population characteristics (eg, enrollment methods, number of participants, special populations, victims of violence, health disparities), regulatory issues mentioned and discussed (eg, regulatory body discussed, challenges addressed), along with conclusions and recommendations for researchers (table 1). Categories within this search included obtaining informed consent, working with legally authorized representatives (LARs), research ethics, research subject protection, understanding of applicable regulatory rules and processes, use of single IRBs for multisite studies, exception from informed consent (EFIC), patient participation, and recruitment. The characteristics of each article were summarized (52 elements) and a narrative synthesis of the results is presented following the PRISMA-ScR guidelines (figure 1).11

Preferred Reporting Items for Systematic Reviews and Meta-Analyses flow diagram of studies for inclusion in a scoping review of human subjects’ protection and regulatory challenges in conducting emergency research.
Study characteristics
Results
Search results
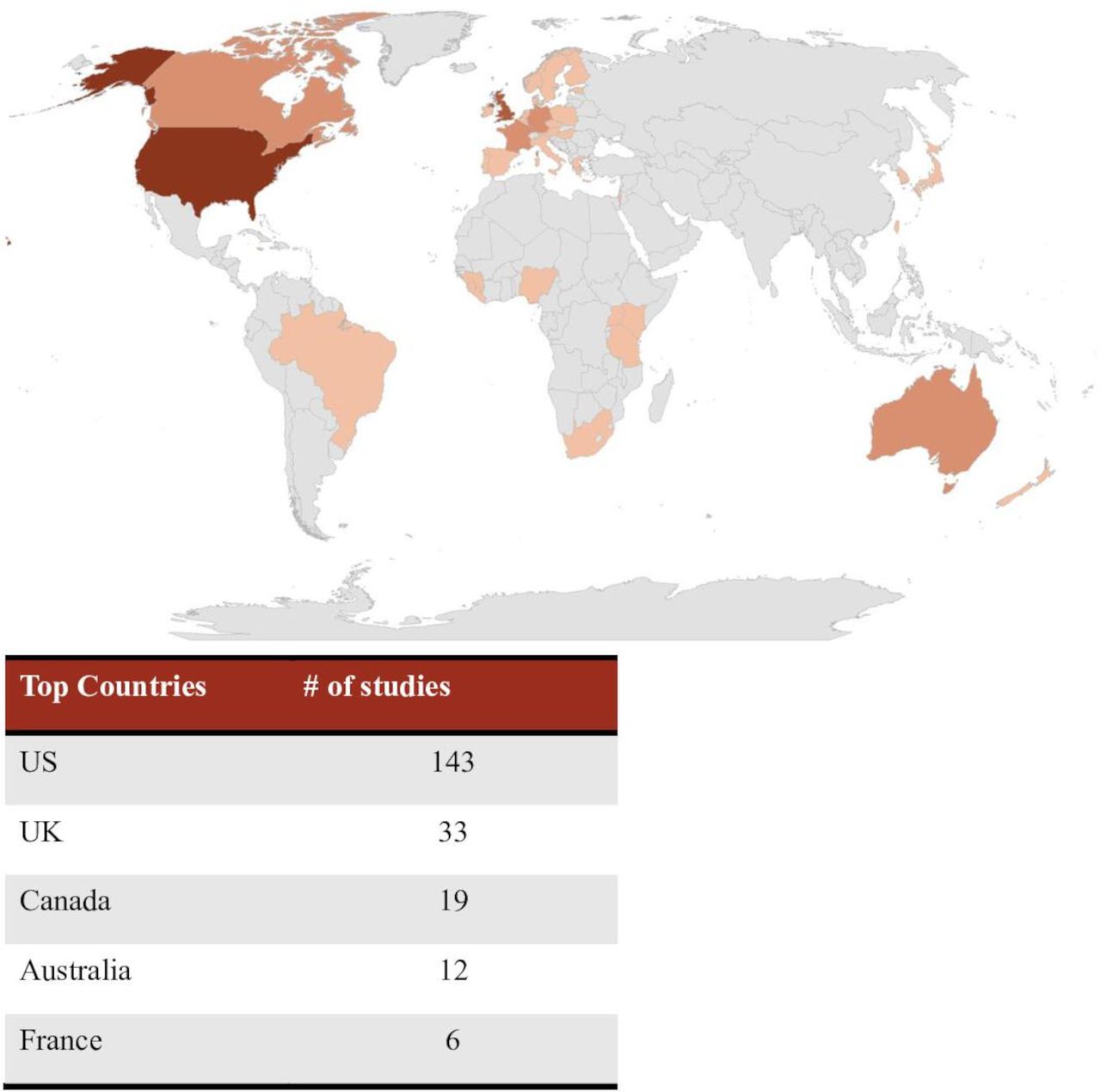
The search returned 2178 original articles for initial screening. Based on the inclusion and exclusion criteria, 289 studies were included in the final data extraction (figure 1). Most articles were from the US (49%), followed by the UK (11%), Canada (7%), Australia (4%), France (2%), Germany (2%), and 21% of articles did not reference a specific country (figure 2). Most articles (31%) were editorial/commentary in nature.

Heat map illustrating the countries in which the studies were conducted.
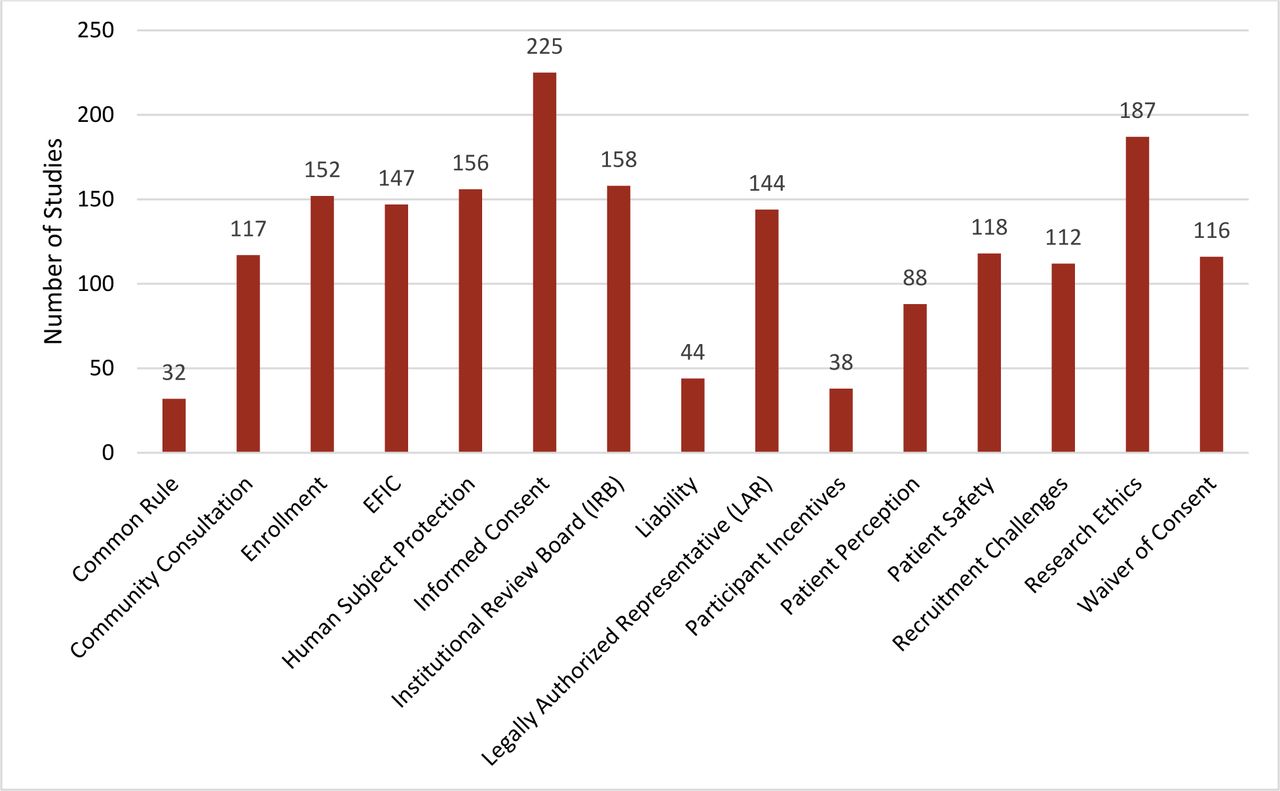
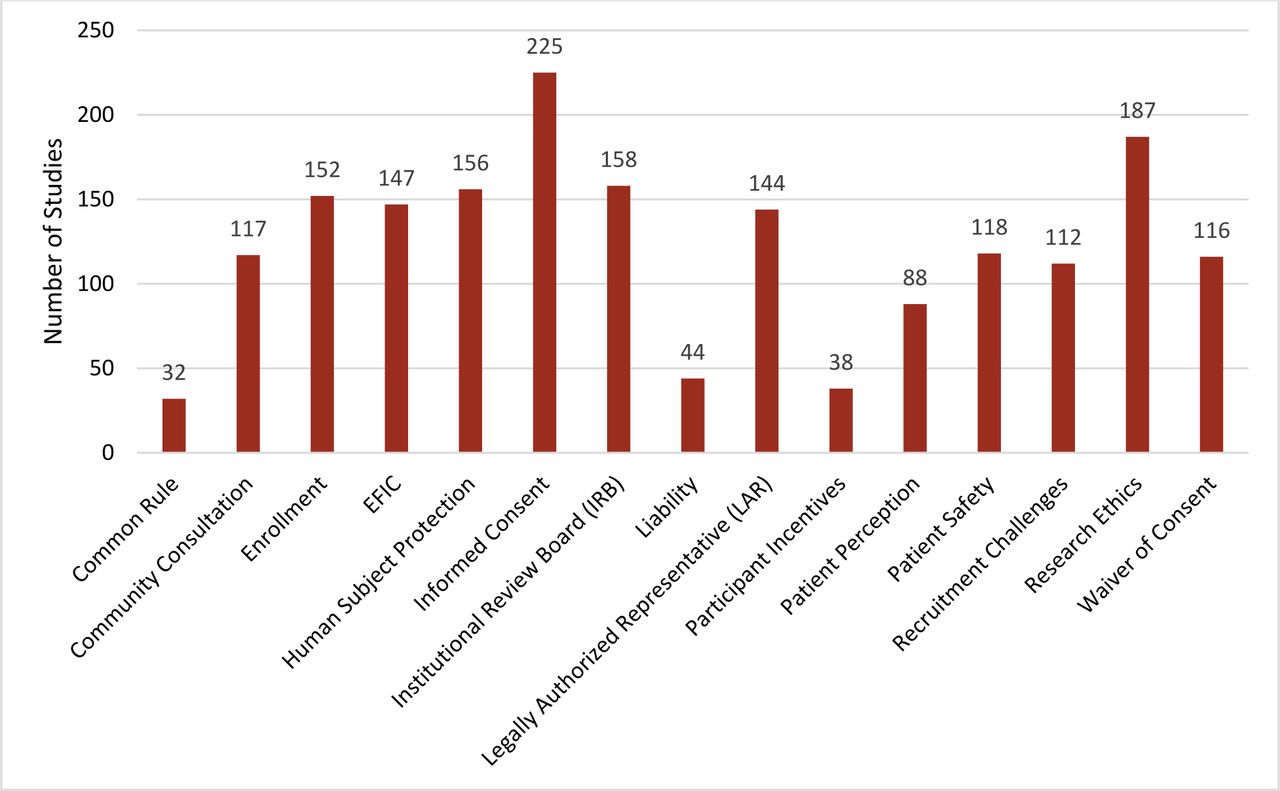
The research team worked with the NTRAP Investigators Group to identify regulatory-related keywords to be used as important categories. Table 2 shows the distribution of the regulatory challenges addressed. Regulatory challenges discussed in the articles were classified into 15 categories (figure 3): informed consent (225; 78%), research ethics (187; 65%), IRB (158; 55%), human subjects protection (156; 54%), enrollment (152; 53%), EFIC (147; 51%), engaging LARs (144; 50%), patient safety (118; 41%), community consultation (117; 40%), waiver of informed consent (WIC) (116; 40%), recruitment challenges (112; 39%), patient perception (88; 30%), liability (44; 15%), participant incentives (38; 13%), and the common rule (32; 11%). We also noted mentions within the articles about challenges specific to research engaging special populations and research during a pandemic or disease outbreak. Based on these results of the scoping review, 15 regulatory challenge topic areas were selected for inclusion in the thematic analysis (table 3). A complete list of challenge statements is mentioned in online supplemental appendix C. The regulatory topic areas below are listed from highest to lowest percentage; yet, researchers will have the ability to review the challenges identified to determine the areas to focus on. Furthermore, challenges that are associated with rules and policies can easily be identified and grouped together to address with the appropriate regulatory agencies.
{kind=link}
{kind=link}
{kind=link}
Regulatory challenges addressed. EFIC, exception from informed consent.
Regulatory challenges addressed
Definitions of regulatory challenges
Informed consent
Informed consent was examined in 78% of the articles. Challenges identified included:
Improving readability of informed consent documents (eg, reducing redundancy, length of forms).
Communicating elements of informed consent in non-overwhelming and understandable ways.
Adequately considering cultural biases when addressing informed consent.
Developing trust and rapport with patients during the informed consent process.
Research ethics
Researchers have a responsibility to protect their patients, provide risks and benefits, and disclose adverse events. Research ethics were discussed in 65% of the articles that cited challenges such as:
Ensuring study efficiencies to minimize patient exposure to potentially ineffective or unsafe therapies.
Ensuring adverse events disclosure to participants.
Ensuring that the patients who are the most likely to benefit from the research have the opportunity to participate.
Determining if regulations for emergency research may threaten public health by impeding advances in life-saving treatments.
Institutional review board
IRBs are formally designated to review and monitor research involving human subjects. Over half of the articles (55%) detailed challenges that researchers face concerning the tasks that are required of their IRBs including:
Using multiple IRBs in a multicenter clinical trial, leading to inconsistent interpretation and consideration of the same protocol at different sites.
Varying implementation of community consultation and public disclosure activities across trial sites as determined by local IRBs.
Encouraging institutions involved in multi-institutional studies to use joint review or reliance on the review of another qualified IRB to avoid duplication of effort.
Working with local IRBs that have little to no experience with EFIC.
Human subjects protection
Protecting patient information, their personal well-being, and interacting with patients are all important when protecting human subjects. Human subjects’ protection was discussed in 54% of articles. Challenges discussed included:
Providing trial information that accommodates the lack of health literacy among large segments of the US population.
Ensuring participants’ concerns and opinions are being heard prior to, during, and after enrollment.
Ensuring that IRBs are adequately overseeing patient safety throughout clinical trials.
Recognizing all potential conflicts of interest, disclosing them properly, and working with the IRB and research team to minimize them.
Enrollment
The ability to determine eligible research participants and enroll them in a research study was discussed in 53% of articles. Specific challenges included:
Ensuring participant enrollment within the recruitment window.
Creating a network of institutions to produce larger patient populations.
Addressing possible impacts of enrollment in more than one trial (co-enrollment).
Developing trust and rapport with patients and families.
Exception from informed consent
EFIC allows patients to be treated as part of research studies without consent under special and rare circumstances. It can only be used in life-threatening emergencies, when there is a possibility for direct benefit to participants, and when consent is not possible. When using EFIC, researchers must ensure that the community is aware of the study and its benefits and opt-out procedures. EFIC was discussed in 51% of articles. Specific challenges included:
Recognizing that some participants will not recall any community consultation or public disclosure efforts before their enrollment.
Effectively communicating with community members regarding an EFIC study and opt-out procedures.
Understanding why community members opt out and providing adequate opt-out opportunities.
Employing adequate opportunities for community members to opt out of EFIC trials.
Legally authorized representative
The use of LARs in which an authorized person may consent on behalf of a participant was discussed in 50% of the articles. Competency, capacity, and comprehension of the LAR were discussed as challenges, including:
Determining who is eligible to be a patient’s LAR as dictated by-laws.
Locating a patient’s LAR in a timely manner.
Ensuring that a LAR knows what the patient’s wishes would be.
Evaluating whether a LAR is competent to provide consent.
Patient safety
Patient safety was discussed in 41% of articles. Key challenges to consider include:
Ensuring that researchers place participants’ welfare ahead of their own interests.
Validating that a researcher’s assertion that the medication, technique, equipment, or system being tested is at least no worse than the current standard of care.
Estimating the amount of psychological and emotional distress that may emerge when asking participants questions about their past traumatic experiences.
Balancing the need for researcher objectivity with the need to decrease the emotional distance between researcher and participant in trauma settings.
Community consultation
Community consultation is a special protection when EFIC is granted for emergency research. Forty percent of articles detailed challenges with the community consultation process including:
Gathering more information on community consultation to better understand the various levels of support, opposition, and uncertainty that are present in the community.
Using patient stakeholders in the development of content for materials and website that will be shared during the community consultation process.
Garnering interest and engaging the community in the consultation process.
Using public disclosure methods to ensure that the target population has a general understanding of the research study.
Waiver of informed consent
Under a WIC, a researcher receives IRB approval to use a person’s personal or health information without actually obtaining consent in order to use that information in a research project (eg, to determine if someone may be eligible for enrollment). WIC was discussed in 40% of articles and the following challenges were cited:
Ensuring that enrollment under WIC only occurs in instances where it is reasonable to believe the patient would typically have consented and the study is not culturally or morally controversial.
Verifying that the magnitude of harm/discomfort anticipated in the research is not greater than encountered in routine medical examination and testing.
Encouraging consistent and rigorous reporting of regulatory prestudy requirements for publications and websites such as ClinicalTrials.gov.
Ensuring that enrollment under WIC only occurs in instances where it is reasonable to believe that the project is not culturally or morally controversial.
Recruitment
The recruitment window often impacts the ability to identify eligible patients quickly and adequately. Recruitment was discussed in 39% of articles, and specific challenges included:
Reducing delays in identifying eligible patients.
Accurately representing the benefits of the study.
Burdening patients and families by trying to recruit during a vulnerable time.
Recruiting patients to more than one study (bombarding the patient).
Patients’ perception
Patients’ perception was identified as a challenge in 30% of the articles, and some examples of challenges cited included:
Engaging community members to determine the acceptability of medical research and resuscitation research in particular.
Fostering patient engagement in the development and conduct of emergency and trauma research.
Minimizing the therapeutic misconception (ie, the tendency of prospective participant to assume that participation improves their chances for a favorable outcome).
Measuring the rate of approval or acceptance of an EFIC study among community members.
Liability
Liability pertaining to the legal risks associated with human subjects’ research was identified in 15% of the articles. Challenges included:
Identifying valid instruments to assess capacity to consent among intoxicated patients.
Obtaining written informed consent in the prehospital transport environment.
Managing liability associated with data repositories and data sharing.
Determining if the potential benefits of a study conducted under a waiver of consent justifies possible infringement on individual rights.
Participant incentives
Another challenge that was addressed in 13% of articles was participant incentives or payments that are made to compensate individuals for participation in research studies. Challenges pertaining to incentives included:
Accurately disclosing the type and size of incentives.
Determining if incentives improve recruitment and retention.
Identifying an appropriate payment or incentive for specific populations.
Selecting incentives that are easy and convenient to use.
Common rule
The common rule, which outlines the criteria and mechanisms for IRB review of human subjects research, was discussed in 11% of articles. Some examples of challenges were:
Ensuring that research complies with federal regulations.
Reducing the administrative burden of research.
Redefining criteria and terminology associated with vulnerable populations.
Furthermore, across all regulatory topic areas, we evaluated articles to determine the specific challenges presented by outbreak-related issues, special populations, and disparities.
Outbreak/COVID-19-related issues
The challenges associated with conducting research during a pandemic or disease outbreak were discussed in 9% of articles. Flexibility is a critical component that emerged as the overall theme in the ever-changing climate characterized by an outbreak or pandemic. Some specific challenges cited were:
Expeditiously reviewing large clinical trial protocols.
Modifying the clinical trial protocol and design based on what is happening during an outbreak.
Maintaining the flexibility to adapt as an outbreak evolves.
Using alternative consent models to feasibly conduct research during a pandemic.
Special population and disparities
Researchers recognize that protecting special participant populations such as pregnant women, prisoners, children, physically or mentally impaired persons, economically or educationally disadvantaged persons, and other vulnerable groups must be a research priority. Thirty-one percent of articles discussed special populations and cited challenges such as:
Communicating the unique risks and benefits that apply to children and special populations.
Ensuring that the enrollment of racial/ethnic minorities is proportionate to the prevalence of the condition being studied.
Addressing mistrust of research investigators by various racial and ethnic groups.
Avoiding the exploitation of vulnerable populations by researchers during emergency situations.
Discussion
Regulatory barriers to trauma and emergency research
Regulations, intended to protect research subjects, impede scientific advancements needed to treat the critically ill and injured. A greater understanding of regulatory barriers will help to ensure that patient safety is maintained at all times.12 Misinterpretation of regulatory requirements causes research teams to not have a full understanding of the intended goal of specific rules, regulations, and policies. Additionally, coordinating clinical studies across multiple sites and IRBs can be time-consuming and inefficient; however, multisite studies are necessary because a single trauma center usually do not have sufficient patient volume to conduct an adequately powered study. A recent report by the Defense Health Board noted, ‘The IRB process is currently fragmented across the Services with different protocol templates, requirements, and methods of implementation’.13 Investigators may be unaware of the unique DoD requirements for the protection of human subjects (eg, second-level review by HRPO)14 and of specific language in consent forms.15 Additional barriers include fears of legal liability16 and misunderstanding of the types and sizes of incentives provided to both participants and researchers engaged in clinical trials.17
Challenges in obtaining consent and enrollment
Preserving the rights and welfare of patients is at the forefront of human subjects protections in trauma research.18 Traditional informed consent indicates that adequate dialogue has occurred between a potential participant and a researcher.19–21 However, previous trauma research demonstrates that severely injured trauma patients can seldom provide consent at the time of injury, and an LAR is often unavailable, in which case EFIC is necessary to recruit a representative sample.22 Furthermore, as the trauma setting is emotionally complex, emergency researchers must be conscientious when approaching emotionally distraught LARs for trial enrollment.23
Implementation of EFIC trials requires a process of community consultation with those who could be potentially enrolled in the trial. Through engagement with community members, researchers are able to measure community members’ perceptions of the proposed research activities. However, different IRBs have varied interpretations of the level of community engagement and support required to move forward with trial enrollment. The logistical challenges in implementing these requirements and the variable approaches have been seen as a barrier to the conduct of EFIC research. Further complicating the issue, there is a unique requirement for a high-level waiver from the Secretary of the Army prior to conducting DoD-funded research using EFIC.
Limitations
Although publications originating from other countries were identified in the review, analysis of regulatory challenges was limited to those pertaining to the USA because the project’s scope is to develop an action plan for the USA. Therefore, this review did not explore regulatory challenges related to conducting international or multinational research. This review also did not detail the limitations in conducting research on military service members. There are special and unique requirements for active-duty service members in participating in research.
Next steps
In the next phase of NTRAP development, CNTR convened a multidisciplinary expert panel to complete an online Delphi survey regarding the importance or impact of the challenge statements identified in the scoping review. The panel reviewed survey results, made recommendations to address the most challenging topics, and outlined strategies to overcome these barriers. The results of the Delphi survey will be submitted for publication in 2023. Additionally, the results will be shared with regulatory bodies and the investigator team will request clarifications on guidance documents. The scoping review and stakeholder survey results will be included in the NTRAP. Including this critical information regarding regulatory challenges will serve to better direct the NTRAP, with the goal of refining the future direction of trauma research. The final step of these activities will be the creation of an investigators’ toolkit for navigating regulatory requirements in trauma and emergency settings research.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
The authors would like to thank Paul Bain, Countway Library, Harvard Medical School, Boston, Massachusetts for his guidance in the development of the scoping review’s search strategy.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Collaborators National Trauma Research Action Plan (NTRAP) Investigators Group: Jeffrey A Bailey, MD baileyja@wustl.edu. Pamela J Bixby, MA Pam@NatTrauma.org. Karen J Brasel, MD brasel@ohsu.edu. Maxwell Braverman, DO Maxwell.Braverman@sluhn.org. Eileen M Bulger, MD ebulger@uw.edu. Zara R Cooper, MD zcooper@bwh.harvard.edu. Todd W Costantini, MD tcostantini@ucsd.edu. James R Ficke, MD jficke1@jhmi.edu. Nicole S Gibran, MD nicoleg@uw.edu. Jonathan I Groner, MD jonathan.groner@nationwidechildrens.org. Adil H Haider, MD, MP Hadil.haider@aku.edu. Bellal A Joseph, MD bjoseph@surgery.arizona.edu. Craig D Newgard, MD newgardc@ohsu.edu. Michelle A Price, PhD Michelle@NatTrauma.org. Deborah M Stein, MD, MPHdstein@som.umaryland.edu.
Contributors Design: CLV, MAP, EMB, National Trauma Research Action Plan (NTRAP) Investigators Group. Data acquisition: CLV, MAP, JPH-E, ANM, AZ, CS, AT. Analysis and interpretation: CLV, ANM, MAP, CAS. Drafting and critical revision: CLV, MAP, ANM, PJB, JPH-E, AZ, CS, AT, EMB, CAS, National Trauma Research Action Plan (NTRAP) Investigators Group.
Funding This work is supported by the US Army Medical Research and Materiel Command under Contract No. W81XWH-18-C-0179.
Disclaimer The views, opinions, and/or findings contained in this manuscript are those of the authors and should not be construed as an official Department of the Army position, policy, or decision unless so designated by other documentation. In the conduct of research where humans are the subjects, the investigators adhered to the policies regarding the protection of human subjects as prescribed by Code Federal Regulations (CFR) Title 45, Volume 1, Part 46; Title 32, Chapter 1, Part 219; and Title 21, Chapter 1, Part 50 (Protection of Human Subjects). This manuscript has been reviewed by the NTRAP Publications Committee for scientific content and consistency of data interpretation with previous NTRAP publications.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.