Article Text

Abstract
Background Septic shock is a public health problem with high mortality. There remains a knowledge gap regarding the optimal resuscitation fluid to improve clinical outcomes, and the underlying mechanism by which fluids exert their effect. Shock-induced endotheliopathy (SHINE) is thought to be a shared pathophysiologic mechanism associated with worsened outcomes in critically ill trauma and sepsis patients. SHINE is characterized by breakdown of the glycocalyx—a network of membrane-bound proteoglycans and glycoproteins that covers the endothelium. This has been associated with capillary leakage and microvascular thrombosis, organ dysfunction, and mortality. Biomarkers of SHINE have been shown to correlate with clinical outcomes in patients with septic shock. Interventions to mitigate SHINE may improve outcomes in patients with septic shock. In surgical/trauma patients with septic shock, initial plasma resuscitation as compared with balanced crystalloid (BC) resuscitation will mitigate biomarkers of SHINE and improve clinical outcomes.
Methods A pilot, single-center randomized controlled trial (RCT) will compare initial plasma to BC resuscitation in surgical and trauma patients with septic shock. Patients will be enrolled based on a Sepsis Screening Score of ≥4 with a suspected source of infection. Patient randomization only occurs if they meet the criteria: (1) hypotension with mean arterial pressure <65 mm Hg, and (2) evidence of hypoperfusion including lactic acid >4 mmol/L, altered mental status or decreased urine output of <0.5 mL/kg in the past hour.
Results The primary outcome is a reduction in serum biomarkers at 6 hours. Secondary outcomes will include clinical outcomes such as intensive care unit-free days, organ dysfunction, and in-hospital mortality.
Discussion This trial will provide insights into the effects of initial plasma resuscitation on SHINE. Furthermore, it will provide unbiased estimates regarding the feasibility, safety, and clinical efficacy of plasma resuscitation in septic shock on which to base subsequent adequately powered multicenter RCTs.
Trail registration number ClinicalTrials.gov (NCT03366220).
- septic shock
- resuscitation fluid
- endothelial dysfunction
- plasma
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Sepsis and septic shock are worldwide public health problems. Although implementation of the evidence-based Surviving Sepsis Guidelines has resulted in improvement in sepsis-related deaths, in-hospital mortality continues to range from 12% to 40%, with an estimated 5.3 million global deaths annually,1 and an estimated healthcare cost of US$14 to $24 billion per year in the USA.2 3 Septic shock survivors often suffer from diminished cognitive function, psychological problems, increased malignancy risk, increased hospitalization rates and long-term damage to organ systems.4 The guidelines provide strong recommendations for initial resuscitation with crystalloids—30 mL/kg within the first 3 hours.5 However, the guidelines state that there is ‘little available evidence from randomized controlled trials (RCT) to support its practice; this is an area in which research is urgently needed.’5 Furthermore, balanced crystalloids (BC) appear to be safer than normal saline, and albumin yields similar outcomes as crystalloids, thus the optimal fluid type is unknown.6
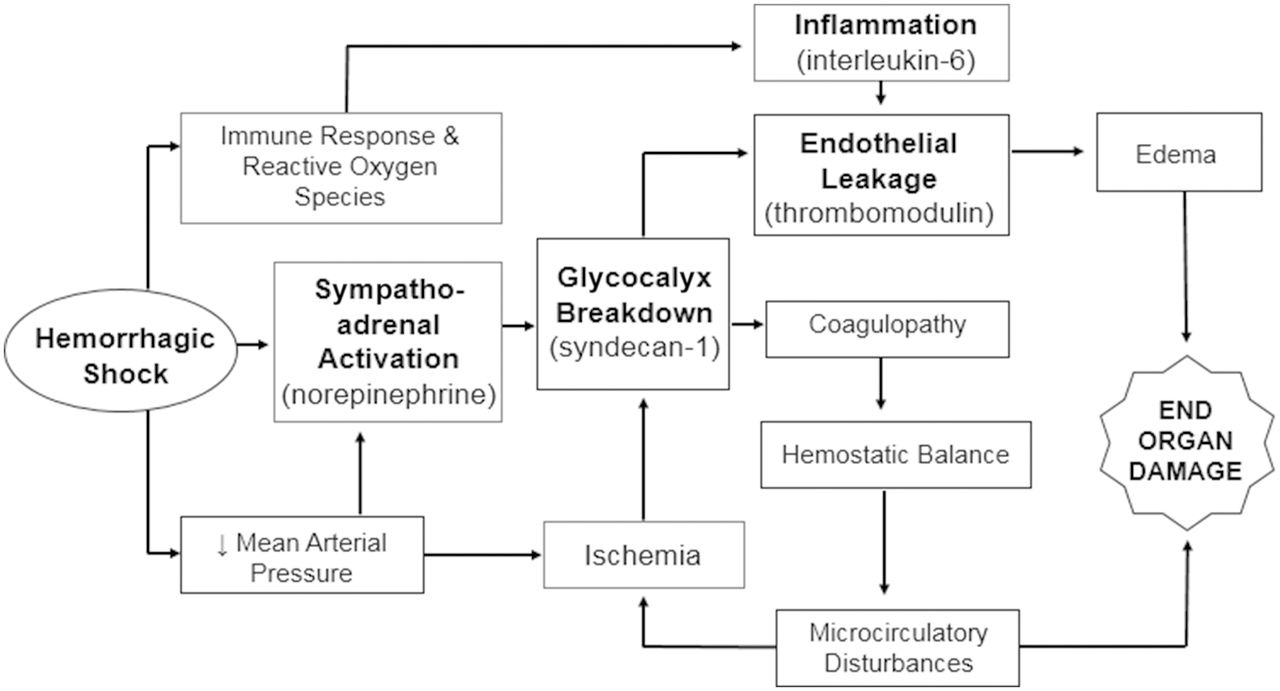
Shock-induced endotheliopathy (SHINE) has been proposed as a mechanism associated with worsened outcomes in patients with critical illness including sepsis.7 Activation of the sympathoadrenal system and release of catecholamines (ie, sepsis or trauma) leads to glycocalyx injury. The glycocalyx consists of a network of membrane-bound proteoglycans and glycoproteins that covers the endothelium (figure 1).8–10 This is associated with capillary leakage and microvascular thrombosis, ultimately resulting in organ dysfunction and increased mortality. Research suggests that endotheliopathy is both a marker and driver of worsened outcome.7 Interventions to prevent, mitigate or treat SHINE may improve outcomes in patients with shock. SHINE biomarkers, such as syndecan-1,11 interleukin-6 (IL-6) and soluble fms-like tyrosine kinase (sFLT-1),10 have been shown to correlate with clinical outcomes in patients with septic shock.
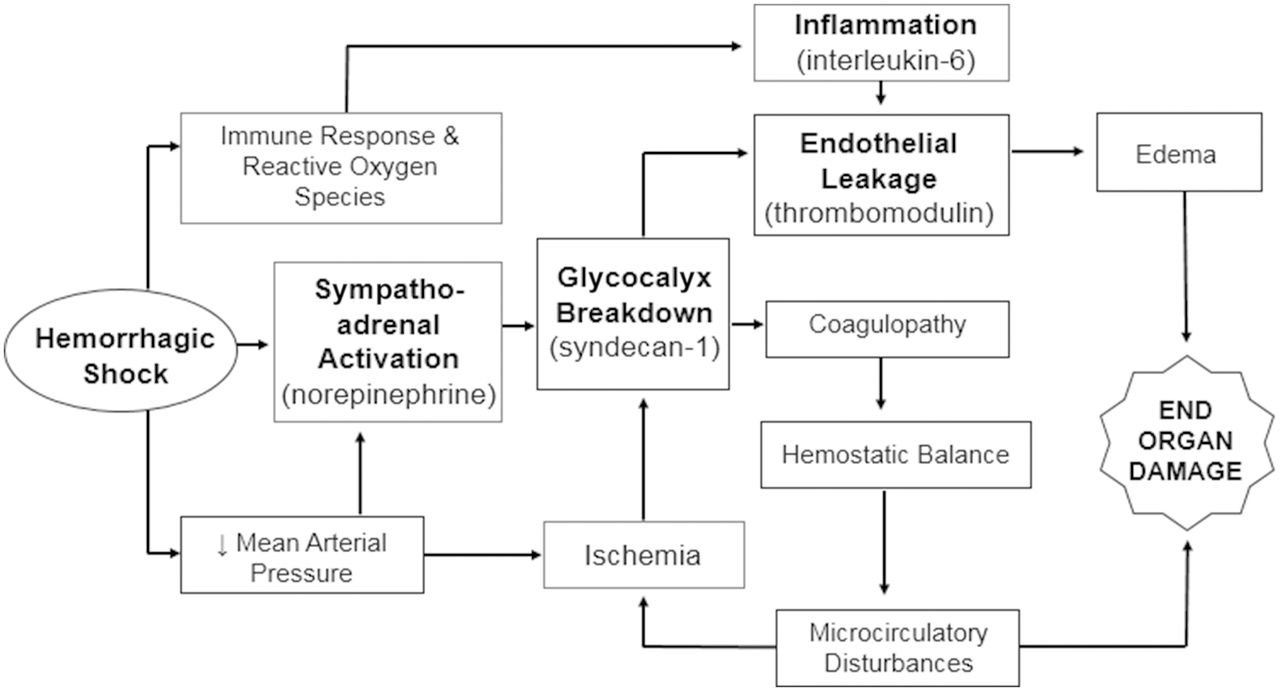
Conceptual model of how septic shock leads to sympathoadrenal activation, inflammation and ischemia, which affects the endothelial glycocalyx, endothelial permeability and hemostatic balance, resulting in edema and microcirculatory disturbances that cause organ dysfunction.
Resuscitation of patients with shock using plasma shows promise as a novel resuscitative strategy. Specifically, plasma may improve outcomes by modulating SHINE as measured by biomarkers of glycocalyx damage (ie, syndecan-1)12 and endothelial injury (ie, sFLT-1, sTM).10 11 Resuscitation with plasma as the primary volume expander in trauma patients has been associated with a reduction in serum biomarkers of endotheliopathy,13 improved survival and decreased morbidity associated with inflammatory and edema-related complications such as acute lung injury and abdominal compartment syndrome.14
We hypothesize that among surgical patients with septic shock, initial resuscitation with plasma versus BCs will attenuate rise in serum biomarkers of endotheliopathy, improve patient outcomes (such as decreased number of intensive care unit (ICU)-free days, decreased morbidity associated with end-organ damage) and reduce in-hospital all-cause mortality.
Methods/design
The trial is a pilot, efficacy, single-center RCT comparing initial plasma with BC resuscitation in surgical patients with septic shock. The trial will investigate modulation of SHINE as a strategy by which outcomes in patients with shock may be improved. The trial will follow the CONSORT (Consolidated Standards of Reporting Trials) guidelines15 and has been registered on ClinicalTrials.gov (NCT03366220).
Setting
The study will take place in the Emergency Department (ED) and 23-bed Shock Trauma Intensive Care Unit (STICU) at Memorial Hermann Hospital-Texas Medical Center, a level I trauma center located in Houston, Texas.
Study population
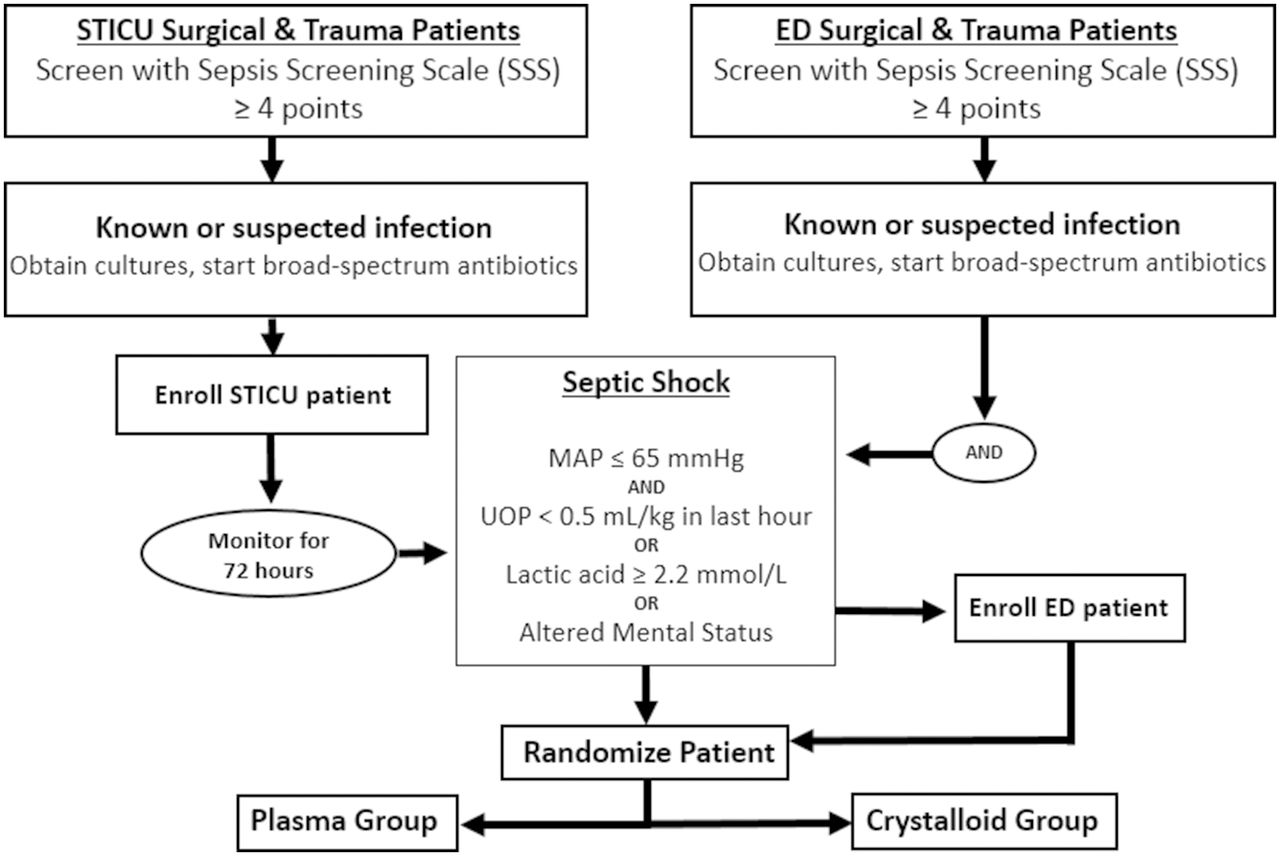
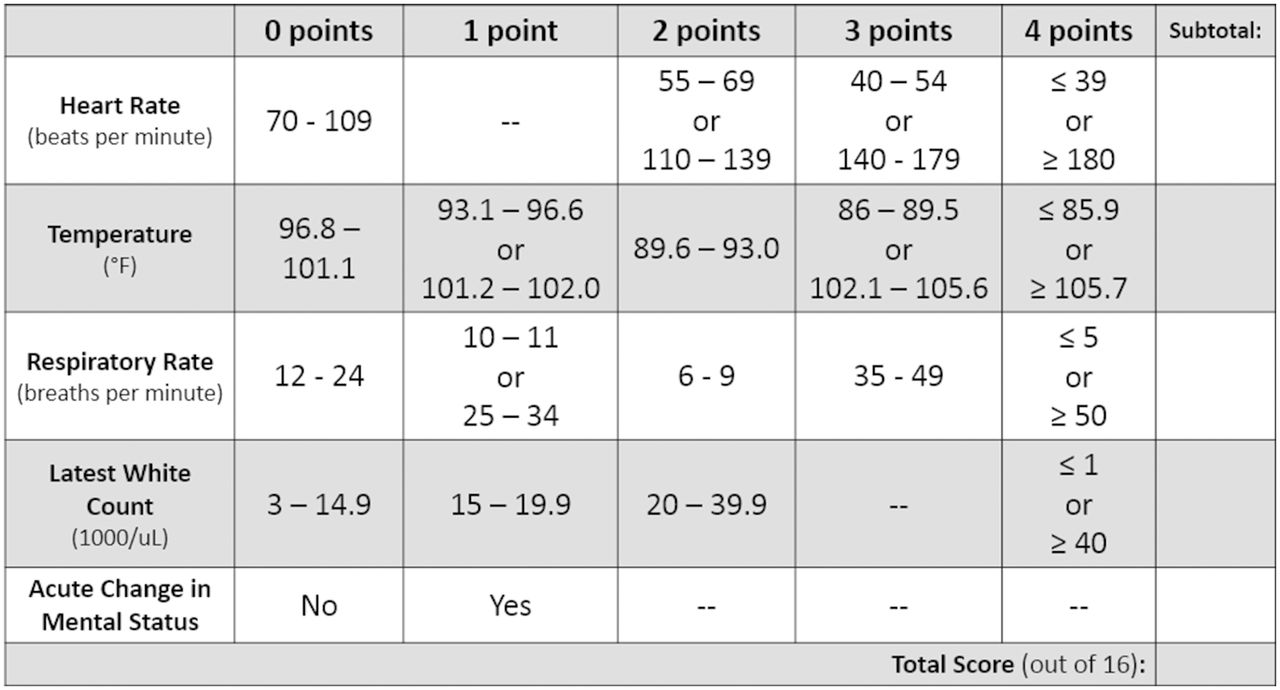
The target population includes critically ill, traumatically injured or surgical patients who have septic shock. Patients who meet the inclusion criteria of ≥18 years old and have a Sepsis Screening Score ≥416 with a suspected source of infection (figure 2) will be eligible for enrollment. Enrolled patients with hypotension with mean arterial pressure <65 mm Hg, and signs of hypoperfusion such as lactic acid >2.2 mmol/L, altered mental status or decreased urine output (<0.5 mL/kg in the past hour) will be randomized to receive either crystalloid or plasma resuscitation. Exclusion criteria are listed in figure 3.
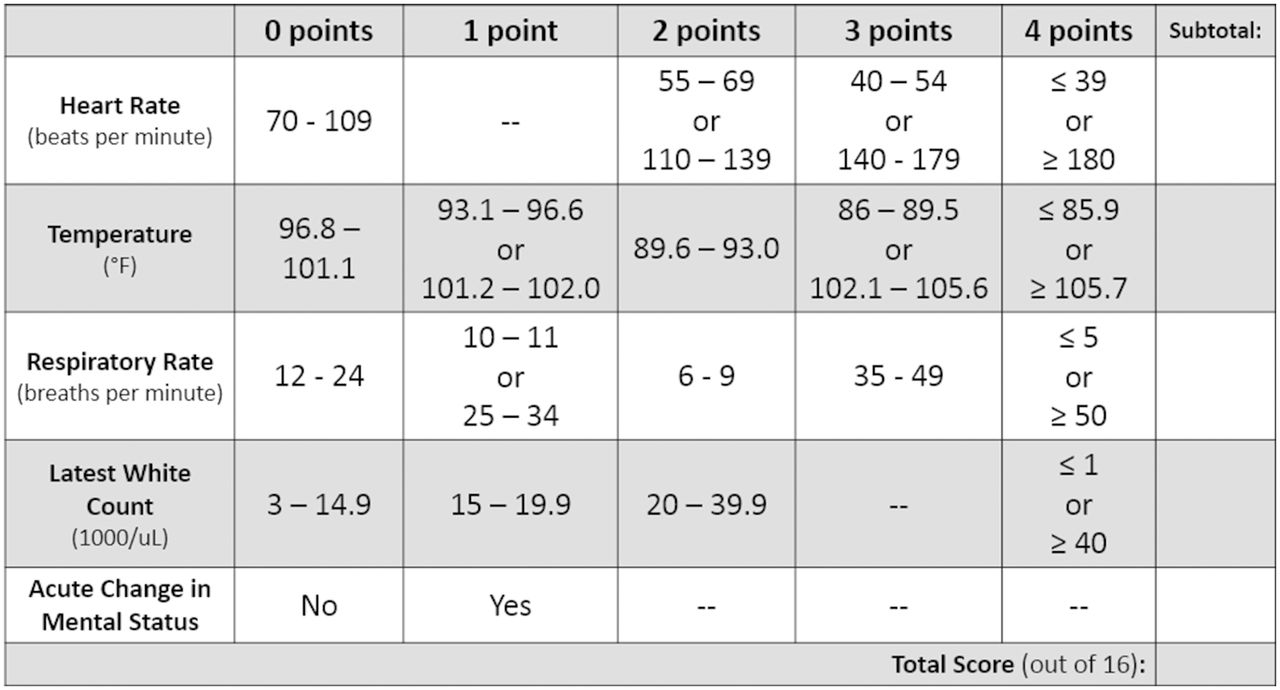
The Sepsis Screening Score. Score of <4 out of 16 has a 96% negative predictive value for sepsis.16

Patient exclusion criteria.
Screening and enrollment
Patient screening and enrollment will take place in the STICU and ED (figure 4). In the STICU, on-call residents and fellows will communicate with research personnel when a patient has suspected sepsis. Once notified, the research team will evaluate potential patients for enrollment eligibility. Enrolled patients in the STICU will be monitored for up to 72 hours (in time for cultures to result) by the research team for development of septic shock. On meeting these criteria for septic shock, patients will be randomized and immediately started on, or switched to (if patient is already receiving fluids) therapy fluid (either plasma or BC). In the ED, research assistants are available 16 hours/day to screen patients who meet the enrollment criteria.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Patient randomization flow sheet from Shock Trauma Intensive Care Unit (STICU) and Emergency Department (ED). MAP, mean arterial pressure; UOP, urine output.
Crystalloid administration may already be under way prior to patient randomization. Patients who have received the entire 30 mL/kg crystalloid dose prior to enrollment are not eligible to participate in this study. Written informed consent will be obtained from subjects or a legally authorized representative. Once consent has been obtained, and patient meets randomization criteria, prior fluids will be exchanged for therapy fluid (either plasma or BC).
Randomization, allocation concealment and blinding
Randomization will occur once the patient meets the criteria. One-to-one allocation will be used for the randomization to either the intervention arm (plasma) or the control arm (BC). Randomization will be performed using a computer-generated random sequence placed in opaque, consecutively numbered, sealed envelopes kept in the locked research office. A research team member is available 24 hours/day to consent and randomize eligible patients. Patients will be stratified by enrollment location (STICU vs. ED) in five blocks of 4 and one block of 6 for a total of 26 patients. The healthcare provider will not be able to be blinded to the study intervention. The outcome assessors will be blinded when feasible, and the laboratory technicians and statisticians will be blinded.
Intervention
Type and screen will be performed to provide donor-matched plasma for subjects randomized to intervention group. Initial resuscitation with plasma will be 10 mL/kg (700 mL in a typical 70 kg adult). Traditional doses of plasma, when used to correct coagulopathy, range from 10 mL/kg to 15 mL/kg.17 Plasma will be administered at a rate of 2 mL to 3 mL/kg/hour (140 mL to 210 mL/hour in a typical 70 kg adult). After the initial dose of plasma has been given, subsequent fluid resuscitation (crystalloid or colloid) will be given at the discretion of the treating clinician. For patients who proceed to the operating room during fluid resuscitation, the anesthesiology team will continue fluid administration per study protocol.
Control
Usual care using BC (Iso-Lyte) only will follow Surviving Sepsis Campaign guidelines. Controls will receive 30 mL/kg (2100 mL in a typical 70 kg adult) of crystalloids within the first 3 hours.5 Crystalloid administration may be terminated before the entire dose has been administered if patients show clinical improvement, or if the treating clinician has concerns about circulatory overload. After the initial dose of crystalloids has been given, subsequent fluid resuscitation (crystalloid or colloid) will be given at the discretion of the treating clinician.
Endpoints of resuscitation: Fluid resuscitation in both the intervention and the control arms will be titrated to serial reassessments of the patient’s volume status at the discretion of the treating clinician. Serial laboratory examinations will be performed per standard of care in the ICU. Additional fluid support after the initial plasma or crystalloid bolus will be given at the discretion of the treating clinician. A research physician will be at bedside to follow patient resuscitation. Plasma administration may be terminated before the entire dose is administered if patients show clinical improvement, or if the treating clinician is concerned for circulatory overload.
Outcomes
The primary outcome is a reduction in serum biomarkers, soluble-thrombomodulin, which is associated with glycocalyx breakdown and endothelial injury at 6 hours after initiation of fluid therapy in the plasma group. Biomarkers will be drawn at 0 and 2 hours at study fluid administration completion, 6, 12 and 24 hours to evaluate their trend in response to plasma versus crystalloid resuscitation. Lactic acid will be drawn per standard of care to guide resuscitation. Standard labs for patients with critical illness will be obtained per ICU protocol.
Secondary outcomes include additional volume of fluid required for resuscitation after initial bolus of study fluid within the first 24 hours of resuscitation, time on vasopressors, time until lactate normalization, ventilator days, ICU-free days and hospital length of stay. Organ dysfunction will be measured including acute lung injury and acute renal failure. Standardized definitions will be used as outlined in the method of operation used in previously conducted trials by our group.18 Potential harms of plasma administration will be assessed. Risk/safety evaluation will be performed at half recruitment by the Data Safety and Monitoring Board, which consists of a general surgeon, pulmonary critical care physician and a statistician. The trial will be terminated early if a series of adverse events attributable to plasma transfusions were to occur.
Sample size calculation and statistical analysis plan
We will enroll 26 patients to obtain unbiased estimates of treatment effect. We chose 26 patients based on the annual average number of patients with septic shock treated in the STICU assuming a 75% enrollment and randomization rate, and the goal of completing enrollment in 1 year. We will calculate estimates of treatment effect and 95% CIs for all measures. We will perform both a per-protocol and an intention-to-treat analysis, and employ both frequentist and Bayesian methods. For the primary outcome and all continuous outcomes, we will use mixed models that include group and time period as covariates with a random subject effect. We will report group differences and 95% CIs from these models. For binary outcomes, a log binomial will be used to estimate relative risks and 95% CIs. HRs and 95% CIs will be reported for time-to-event comparisons. For the Bayesian analysis, neutral conservative priors will be used to estimate the probability of both benefits and harms, as defined by our primary and secondary outcomes.
Discussion
Although there is biological rationale to support a restorative effect of plasma on the vascular endothelium and on SHINE, there have been no randomized trials in humans to date comparing plasma with BC in fluid resuscitation for septic shock. This pilot trial will be the first to test the hypothesis that plasma can modulate SHINE, as evidenced by a decrease in biomarkers of glycocalyx breakdown and endothelial injury, and improve outcomes in trauma and surgical patients with septic shock. Although the trial is underpowered to identify a reduction in mortality, it will provide the least biased estimate of treatment effect on which to plan further multicenter trials. Furthermore, this study will provide insights regarding the mechanism by which fluids can influence outcomes in septic shock.
Animal models of sepsis and a prospective substudy of an RCT have shown that resuscitation with plasma is associated with decreased levels of syndecan-1, which may reflect restoration of endothelial integrity.19–21 Plasma resuscitation in a rat model of sepsis demonstrated attenuation of inflammatory markers (IL-6), endothelial injury biomarkers (syndecan-1) and catecholamines (norepinephrine); significantly reduced pulmonary edema as measured by wet-to-dry weight ratios; and improved 48 hours of survival as compared with normal saline.22 Although the results of plasma resuscitation in animal models of septic shock appear promising, an RCT is needed to establish whether plasma resuscitation for septic shock is safe and confers similar benefits in humans.
There are several limitations to this study, with the first being the small sample size. Given that there have been no prior studies on which to base a power calculation. Use of Bayesian analyses to complement traditional frequentist analyses will allow estimates of the probability of benefit of plasma. A second limitation is that the primary outcome is based on SHINE biomarkers, which are a surrogate endpoint. The promising effects of plasma on SHINE biomarkers seen in vitro and in animal models may not be replicable in humans. Moreover, changes in biomarkers may not correlate with clinical outcomes. Larger pragmatic trials are planned if the results of the pilot trial suggest potential benefit with plasma. A third limitation of the study is the heterogeneity of patients. ED patients are likely to have community-acquired sepsis whereas STICU patients have hospital-acquired sepsis, which prior studies have shown to have different clinical outcomes.23 We chose to incorporate both STICU and ED patients to capture a greater number of eligible patients. Despite community-acquired and hospital-acquired sepsis being different clinical entities, the current treatment standard for both populations remains the same. To account for this heterogeneity, we will stratify by enrollment location. By not limiting our patient population, the study’s outcome will be more generalizable to the surgical and trauma septic shock populations.
In summary, this study will be the first pilot RCT that compares plasma versus BC in the initial fluid resuscitation of surgical or trauma patients with septic shock. Results from this study will be hypothesis generating, and may be used to inform future, multicenter RCTs.
References
Footnotes
Contributors All authors included in the article contributed to the conception, design, implementation of the trial and drafting of the article.
Funding SW is supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM085232.
Disclaimer The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Advancing Translational Sciences or the National Institutes of Health.
Competing interests None declared.
Patient consent Not required.
Ethics approval This study has been approved by our institutional review board. Committee for the Protection of Human Subjects (HSC-MS-17-0714).
Provenance and peer review Not commissioned; internally peer reviewed.