Article Text

Statistics from Altmetric.com
- CCU, coronary care unit
- MI, myocardial infarction
- MR, mitral regurgitation
- PCI, percutaneous coronary intervention
- PTCA, percutaneous transluminal coronary angioplasty
- STEMI, ST-elevation myocardial infarction, UA/NSTEMI, unstable angina/non-ST-elevation myocardial infarction
The clinical diagnosis of myocardial infarction (MI) relies on symptoms, electrocardiographic findings, and biochemical markers (troponin, serum creatine kinase, creatine kinase-MB).1,2 Acute ischaemic syndromes are now classified as unstable angina/non-ST-elevation MI (UA/NSTEMI) and acute ischaemic syndromes with ST-elevation MI (STEMI).1,2 The new diagnostic criteria and markers are leading to increased proportions3 of acute ischaemic syndromes being recognised as acute MI. Obviously, elevated troponin concentrations are not, by themselves, synonymous with acute MI and can occur in a variety of cardiac and non-cardiac disorders (for example, sepsis or septic shock, pulmonary embolism, acute exacerbation of chronic obstructive pulmonary disease).4 Therefore, the diagnosis of acute MI relies on the combination of all clinical and biochemical tools, each providing its own diagnostic contribution.
The pathological hallmark of acute MI is coagulative necrosis of the myocardium. All recent advances in the definition, diagnostic work-up and treatment of MI are essential to perform an informative pathological investigation. In fatal MI, the pathological study must be performed at the appropriate technical and interpretative level to confirm, extend and improve information useful for the clinical understanding of the event (why one infarction proves fatal while other clinically similar MIs are not) and, eventually, contribute towards improving knowledge that may help future research in the MI setting.
PATHOLOGY
The pathological diagnosis of MI relies on the identification of coagulative necrosis in the myocardium, or of repairing features according to the “age” of the MI,5 or, if death occurred before the time necessary for coagulative necrosis to become visible at routine histopathology, on the detection of occlusive coronary thrombosis of an epicardial coronary artery (International classification of diseases, 9th revision (ICD-9) classification 410, 411). When coronary thrombosis is not detected at autopsy in individuals with MI who did not receive reperfusion, plaque complications such as rupture and haemorrhage can be considered the potential substrate of an acute thrombotic event that spontaneously thrombolysed. In less than 5% of cases, MI is reported as not being associated with coronary atherosclerotic plaques. Coronary spasm (toxic,6 drug-induced (Kounis syndrome)7 or associated with systemic disease8), coronary emboli, and myocardial bridges9 have been considered as exceptional causes of MI; for these coronary substrates, the pathologic identification of the culprit lesion may be difficult. Cases with clinically diagnosed MI in which neither coagulative necrosis nor acute events in the culprit plaque are found at autopsy are exceptional.
Most patients with acute MI who are admitted to coronary care units (CCUs) and coronary interventional labs shortly after the onset of the ischaemia have a favourable prognosis.10 In the modern cardiology setting, fatal MIs are usually those occurring out of the hospital, or are seen in patients who came late to the CCU, did not receive appropriate treatments, or died suddenly from life-threatening arrhythmias.10
With respect to transmural versus subendocardial MI, the recent identification of small intramural foci of coagulative necrosis, clinically recognised with the additional information derived from troponin measurements (fig 1),1 indicates the need for modified investigation protocols at autopsy with extensive search for microfoci of necrosis in multiple myocardial samples. These MIs are unlikely to be fatal unless the acute ischaemia triggers life-threatening arrhythmias and, in any case, the corresponding clinical phenotype should be UA/NSTEMI.

Small foci of coagulative necrosis can be recognised on haematoxylin and eosin (H&E) stained sections: ischaemic myocytes typically show the hypereosinophilia that characterises early phases of coagulative necrosis. (A) The ischaemic myocytes are located in the left side of the panel; (B) the ischaemic myocytes are positioned bottom left; (C) low magnification view showing a small area of acute myocardial infarction in which granulocyte infiltration is clearly visible among the myocytes showing coagulative necrosis (squared area and (D), inset at higher magnification). The front of the myocardial ischaemia is in the top half of the figure.
PRE-INTERVENTIONAL AND PRE-THROMBOLYTIC ERA
Myocardium
Non-reperfused MI shows typical ischaemic coagulative necrosis.5 During the first 30–40 minutes of ischaemia, the changes are visible only at electron microscopy and are reversible. The macroscopic appearance depends on the interval of time between the onset of MI and death. A macroscopic early diagnosis (few hours from onset) relies on the immersion of the infarcted myocardium in a solution of triphenyltetrazolium chloride. This histochemical stain imparts a brick-red stain to the non-infarcted area preserving the dehydrogenase enzymes. From 12–24 hours the myocardium appears as dark mottling; from days 1–3, the mottling is centred by a yellow-tan core; from days 3–7 the central yellow-tan softening area is surrounded by hyperaemic borders; from days 7–10, the infarction area is yellow-tan and soft, and the margins are red-tan and depressed; from 10–14 days, the borders assume a red-grey colour; from 2–8 weeks the scar starts to develop from the periphery to the centre; after the second month, the scarring process should be completed.
Although the microscopic appearance before 12 hours is poorly informative, hypereosinophilic changes of the myocyte sarcoplasms are present before neutrophilic infiltrates (fig 1A,B). The so-called “waviness” may be seen at the border of the ischaemic MI. Isolated myocyte waviness (without other findings such as hypereosinophilia of the sarcoplasms or contraction bands and coagulative necrosis) do not have diagnostic value. Focal waviness of single myocytes or groups of these cells can be seen in hearts of patients who died from proven non-cardiac causes; they constitute the morphologic expression of terminal changes in pre-agonic and agonic phases. The lack of significance of isolated myocyte waviness has been experimentally demonstrated.11
After 12 hours, coagulative necrosis starts and progresses with loss of the nuclei (days 1–3), neutrophilic infiltration (early days 1–3) (fig 1C,D, fig 2A,B), myocyte fragmentation (days 3–7) and early phagocytosis at the border of the MI (days 3–7) after the first week; the granulation tissue progresses and evolves through loose (week 2) and progressively dense collagen deposition (from 3–8 weeks) and scar that is completed by the second month. After that date, the scar becomes acellular and collagen appears dense and compact.5 The above time intervals indicate the onset and peaks of the features but do not reflect the ending. In large transmural MI, layers of necrotic myocytes can be observed after intervals longer than two months.
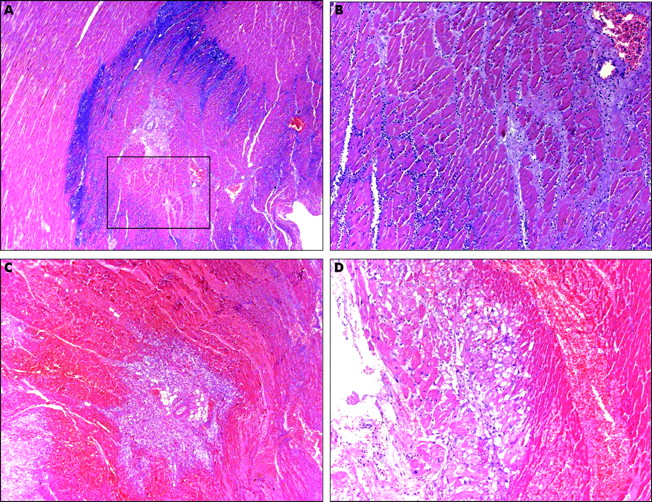
(A and B) Typical, non-reperfused myocardial infarction: the basophilic areas indicate the front of granulocyte infiltration. In non-reperfused MI the repair starts at the borders of the MI and the front progresses from the periphery to the centre. (C and D) Typical reperfusion pattern of a consolidated myocardial infarction: note the extensive haemorrhagic invasion of the myocardium with coagulative necrosis and the absence of haemorrhage in subendocardial layers with morphologically “viable” myocytes.
Coronary arteries
A culprit plaque with acute thrombosis is found at autopsy in more than 90%1,2 of patients who have died from MI and were not treated with either thrombolysis or percutaneous transluminal coronary angioplasty (PTCA). The plaque substrate for thrombosis is rupture in about 75% of the cases and erosion in a minority of cases,12 mostly women and smokers.13 The typical culprit lesion is a large atherosclerotic plaque with cap ulceration and superimposed acute thrombosis. The acute thrombus is red, with a small platelet-rich small head, a fibrin- and red cell-rich body, and a red cell-rich tail.14
POST-THROMBOLYTIC AND POST-PTCA ERA
Reperfusion in MI restores the coronary flow interrupted by the acute coronary event. It can be obtained using thrombolysis or mechanical interventions such as PTCA with or without stenting. The greatest effectiveness is obtained with PTCA which dramatically modifies the natural history of MI and is now available in nearly all tertiary cardiologic centres in Europe.2
Thrombolysis and percutaneous coronary interventions (PCI) with or without stenting is usually performed when the interval between the onset of symptoms and opening of the culprit coronary artery is less than 12 hours (the gold standard is six hours, while the benefit derived from reperfusion between 12–24 hours is debatable). Guidelines for STEMI indicate 12 hours after onset of symptoms, and then distinguish the indications on the basis of the presence or absence of a PCI centre in the hospital. In hospitals where a PCI centre is active, all patients with STEMI should undergo primary PCI. If the interval between onset of symptoms and arrival at a hospital without a PCI centre is between 3–12 hours, the patient should be immediately transferred to a hospital with an active PCI centre. If the interval is < 3 hours, then thrombolysis can be performed.1,2
Myocardium
Reperfusion strategies are introduced in the cardiopathological setting for the so-called reperfusion-associated pathologies, whose clinical manifestations include arrhythmias and prolonged ischaemic dysfunction, the pathological evidence for which includes myocardial haemorrhage with contraction bands, myocyte reperfusion injury distinct from, and additional to, coagulative necrosis, and small vessel damage. Contraction bands are seen in irreversibly injured myocytes: their morphology is characterised by intensely eosinophilic transverse bands comprising closely packed hypercontracted sarcomeres. The macroscopic appearance of reperfused MI is typically haemorrhagic. Microscopic examination of reperfused infarction areas shows myocytes with coagulative necrosis surrounded by red cell infiltration (fig 2C,D), contraction band necrosis and small vessels which are either damaged or showing small thrombo- or athero-emboli. Small vessel damage may further worsen the haemorrhagic invasion of the myocardium and leads to endothelial cell swelling which is potentially occlusive (especially at the capillary level), thus preventing local reperfusion of ischaemic myocardium. This phenomenon is known as no-reflow.15,16 If reperfusion is done before irreversible necrosis, the blood flow restoration of the area at risk may rescue the entire ischaemic myocardium. Alternatively, the rescued area is proportional to the interval elapsed between onset of ischaemia and blood flow restoration. Scars of reperfused MI show more angiogenesis processes than non-reperfused MI. In the majority of cases, the result of reperfusion is a limitation of the infarct area and size, with improvement of short and long term function and prolonged survival.2
Coronary arteries
In reperfused MI the culprit lesion is expected to be patent: it may show ulceration with haemorrhagic invasion of the core or mural thrombus layered over the plaque ulceration. Pultaceous material and thrombotic fragments may reach the small vessels of the area around the culprit vessel.17 This deleterious consequence of infarct-related artery reperfusion can be addressed by the upstream use of glycoprotein IIb/IIIa inhibitors, which were found to improve microcirculatory function and clinical outcome.18 Alternatively, new filters or aspirating devices are being used in clinical practice to collect or suck up both plaque and thrombus derived fragments in order to limit small vessel impairment. However, these devices were not found to sufficiently antagonise the no-reflow phenomenon and improve clinical results.19
MYOCARDIAL INFARCTION COMPLICATIONS
Acute pulmonary oedema
Pulmonary oedema is associated with a 20–40% 30-day mortality rate, even in the fibrinolytic era.20 Pulmonary oedema may occur as an acute event with the onset of STEMI or reinfarction, or as the culmination of slowly progressive congestive heart failure during the first days after MI.
Heart rupture
The main risk factors for heart rupture include longstanding hypertension, female sex, advanced age, and no history of prior infarction.21,22
-
Left ventricular free-wall rupture—Cardiac rupture occurs in 1–6% of all patients admitted with STEMI. The frequency of cardiac rupture shows two peaks: one early within 24 hours, and one late from 3–5 days after STEMI. Risk factors for cardiac rupture include: first MI, anterior infarction, old age, female sex, hypertension during the acute phase of STEMI, lack of prior angina and MI, lack of collaterals, Q waves on the ECG, symptoms of pericarditis, peak MB creatine kinase > 150 IU/l, intake of corticosteroids or non-steroidal anti-inflammatory drugs, and fibrinolytic therapy more than 14 hours after onset of symptoms.21,22 The most important determinants in preventing rupture are successful early reperfusion and the presence of collateral circulation.21,22
-
Ventricular septal rupture—During the reperfusion era the frequency of acute rupture of the interventricular septum has declined. It occurs in less than 1% of patients with STEMI.23 In patients treated with fibrinolytic therapy, the highest risk is within the first 24 hours after MI. The rupture site can rapidly expand and cause sudden haemodynamic collapse, even in patients who appear to be clinically stable with normal left ventricular function.
-
Papillary muscle rupture—Papillary muscle rupture occurs in less than 1% of cases. The diagnosis is made on the basis of clinical and imaging findings.
Left ventricular aneurysm
Aneurysm after STEMI usually occurs in the left anterior wall, in association with left anterior descending occlusion and a wide infarcted area. Patients with STEMI treated with fibrinolytic therapy and a patent infarct-related artery have a significantly reduced incidence of left ventricular aneurysm compared with those who do not (7.2% v 18.8%).24
Ventricular pseudoaneurysm
Ventricular pseudoaneurysm is a rare complication. It occurs as a consequence of rupture of the ventricular free wall and is contained by overlying, adherent pericardium, producing what has been termed a “false aneurysm or pseudoaneurysm” of the left ventricle. The pathologic features depend on the interval of time elapsed from onset of MI and death and from the extent of haemorrhage between the pericardium and the myocardial wall. The myocardial wall shows interruption or fissuring. The myocardial changes include coagulative necrosis with or without reperfusion pattern, according to the administered treatments.25 Most pseudoaneurysms are formed within seven days after an AMI, only exceptionally forming later.
Arrhythmias
Cardiac arrhythmias are common in patients with STEMI and occur most frequently early after the development of symptoms. The mechanisms for ventricular tachyarrhythmia include loss of transmembrane resting potential, re-entrant mechanisms due to dispersion of refractoriness in the border zones between infracted and non-ischaemic tissues, and the development of foci of enhanced automaticity.26 Reperfusion arrhythmias likely involve washout of toxic metabolites and various ions such as lactate and potassium.26
Lethal arrhythmias/sudden death
Ventricular arrhythmias are one of the most frequent causes of death in non-hospitalised patients with acute MI.27 They are the most common form of sudden ischaemic death.
Cardiogenic shock
Cardiogenic shock in patients with STEMI is commonly (75%) caused by extensive left ventricular dysfunction.28 Other relevant causes include mechanical complications (acute severe mitral regurgitation, ventricular septal rupture, and subacute free-wall rupture with tamponade). Cardiogenic shock may be mimicked by aortic dissection and haemorrhagic shock.
Mitral regurgitation
After STEMI, mitral regurgitation (MR) may occur as a result of infarction of the papillary muscle, infarction involving the lateral wall, large infarction with left ventricular dilation, and displacement/dysalignment of the papillary muscle. Severe MR with cardiogenic shock has a poor prognosis. In the SHOCK trial registry, approximately 10% of patients with shock presented with severe MR (overall hospital mortality 55%).28 When severe MR is caused by infarction of the papillary muscle and wall, the area of infarction tends to be less extensive than in patients in whom the MR is caused by papillary displacement/dysalignment and severe left ventricular dysfunction. The presence of acute pulmonary oedema or cardiogenic shock in posterior/posterolateral STEMI should point to the possibility of acute MR caused by papillary muscle rupture.
Pericarditis
Pericarditis occurs in transmural STEMI involving the full thickness of the myocardial wall to the epicardium. Patients with pericarditis have larger infarcts, lower ejection fraction and higher incidence of congestive heart failure. Pericarditis may appear up to several weeks after STEMI. The Dressler syndrome (post-MI syndrome) has essentially disappeared in the reperfusion era.29
Acute pulmonary haemorrhage
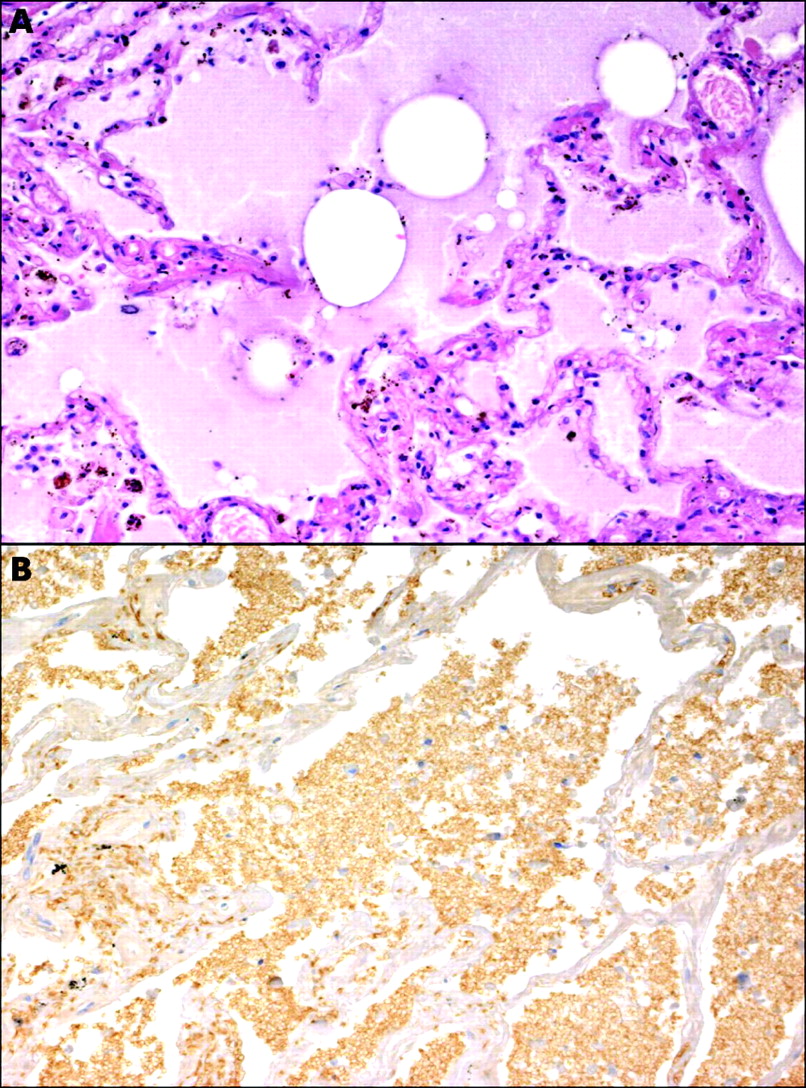
This is a rare complication that may occur in patients who undergo primary PTCA and are treated with glycoprotein IIb/IIIa inhibitors.30,31 When it occurs, it is difficult and costly to treat and may result in death. The pathologic diagnosis is essential to confirm the alveolar invasion by red blood cells (fig 3A,B). Bleeding complications are higher in women than in men. In pooled analysis of the results from EPIC, EPILOG and EPISTENT, major bleeding rates were 3% and 1.3% (p = 0.004) and minor bleeding rates were 6.7% and 2.2% (p < 0.001) in women and men, respectively. Rare intracranial and gastrointestinal haemorrhages have also been reported.32
{kind=link}
{kind=link}
{kind=link}
Light micrographs showing (A) typical pulmonary oedema (H&E stain) versus (B) pulmonary haemorrhage in a patient who died of acute adult-type respiratory distress related to abciximab (peroxidase-antiperoxidase; anti-glycoforin A immunostain).
CONCLUSIONS
The pathology of MI in the post-interventional era includes specific features mostly resulting from the reperfusion of necrotic myocardium. The contribution of the pathologic study should add information to the clinical data and should match new sensitive diagnostic markers. The complication scenario is also modified: prevalence and evolution are significantly different in non-reperfused and reperfused MI.
REFERENCES
Footnotes
-
Supported by grants: Ricerche Finalizzate granted by the Ministry of Health to the IRCCS Policlinico San Matteo, Pavia, Italy
-
Published Online First 18 April 2006