Article Text

Statistics from Altmetric.com
Surgical dilemma
A 58-year-old man with a history of obesity status post Roux-en-Y gastric bypass and chronic lymphocytic leukemia (CLL) with aggressive features in complete remission 10 months after bendamustine and rituximab chemotherapy presented with neurological decline. He previously was a high-functioning information technology expert who 3 weeks earlier began having difficulty with fine movement when using his mobile device. He presented at a local hospital with new-onset dysarthria and word-finding difficulties, prompting admission. He underwent extensive neurological workup, which was negative. He was given an empiric trial of steroids due to suspicion of a neurological paraneoplastic syndrome and had minimal improvement. Cross-sectional imaging of the chest, abdomen, and pelvis showed splenomegaly, with no other lesions or lymphadenopathy. He was transferred to our referral center for further management.
He presented with white blood cell count of 3.1, hemoglobin of 11.5 g/L, and platelet count of 53,000/uL. An fluorodeoxyglucose (FDG)-positron emission tomography (PET) scan was notable only for high metabolic activity in the superior pole of the spleen, concerning for recurrent lymphoma (figure 1). We suspected transformation of CLL into diffuse large B cell lymphoma (DLBCL) (ie, Richter’s syndrome) with accompanying paraneoplastic manifestations. On initial evaluation by the surgical team, he was alert and oriented but had expressive aphasia and word-finding difficulties. On re-evaluation in the afternoon, he was not speaking and no longer following commands. His platelet count was low and minimally responsive to transfusion.

FDG-PET imaging reveals an avid lesion in the anterior superior pole of the spleen, concerning for recurrent malignant disease. There is no evidence of other lymphadenopathy and the bone marrow did not show abnormal uptake in this or other images.
What would you do?
Total splenectomy
Partial splenectomy
Bone marrow biopsy
Continue steroids and attempt to initiate chemotherapy
What we did and why
A. Total splenectomy.
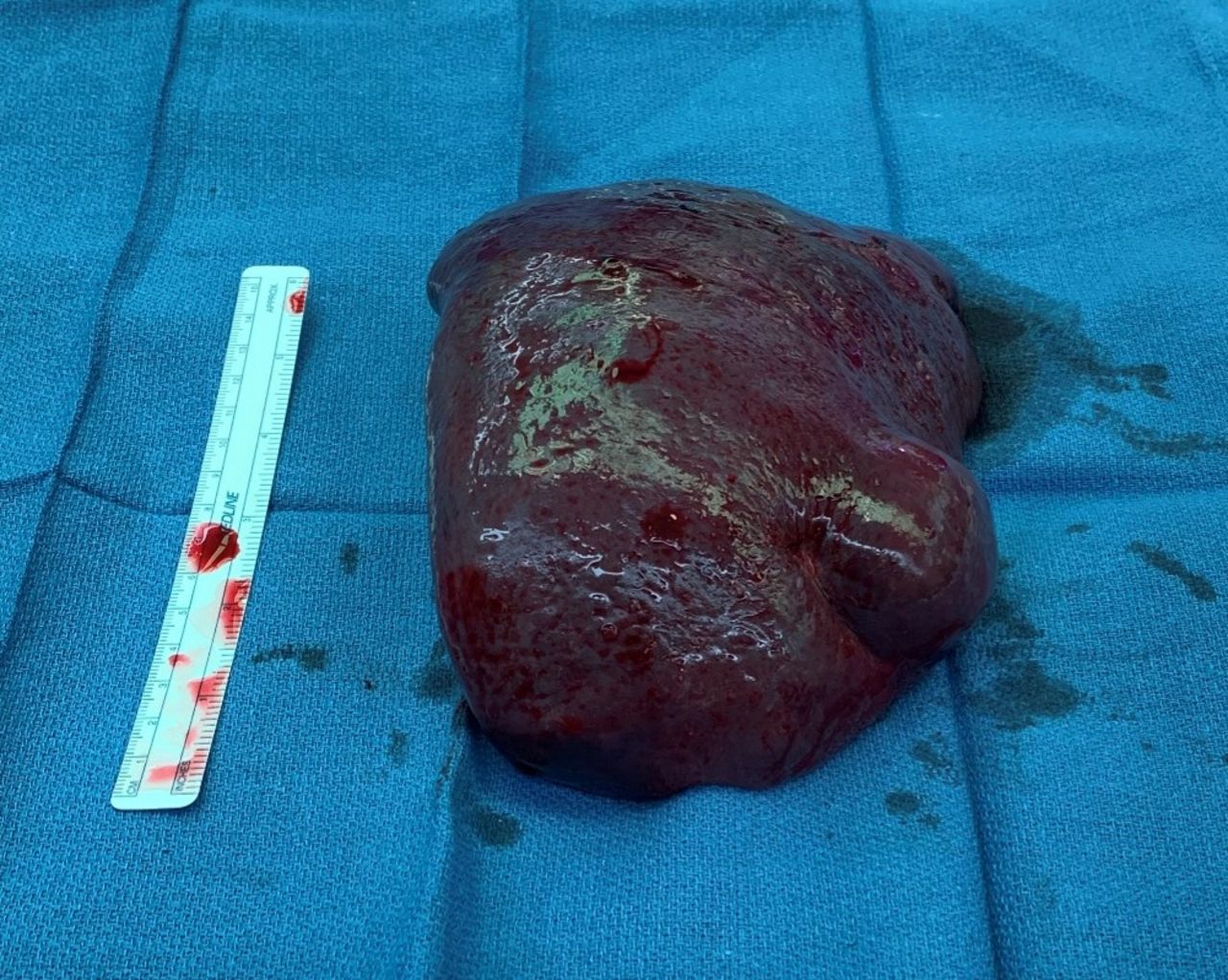
In consultation with the oncology team, the patient was taken for a laparoscopic hand-assisted splenectomy. Additional steroids were given the evening prior to surgery and platelets were transfused in the operating room. Given concerns for extranodal lymphoma, the spleen was not morselized but instead removed intact through the hand port to preserve the architecture for pathological study (figure 2).
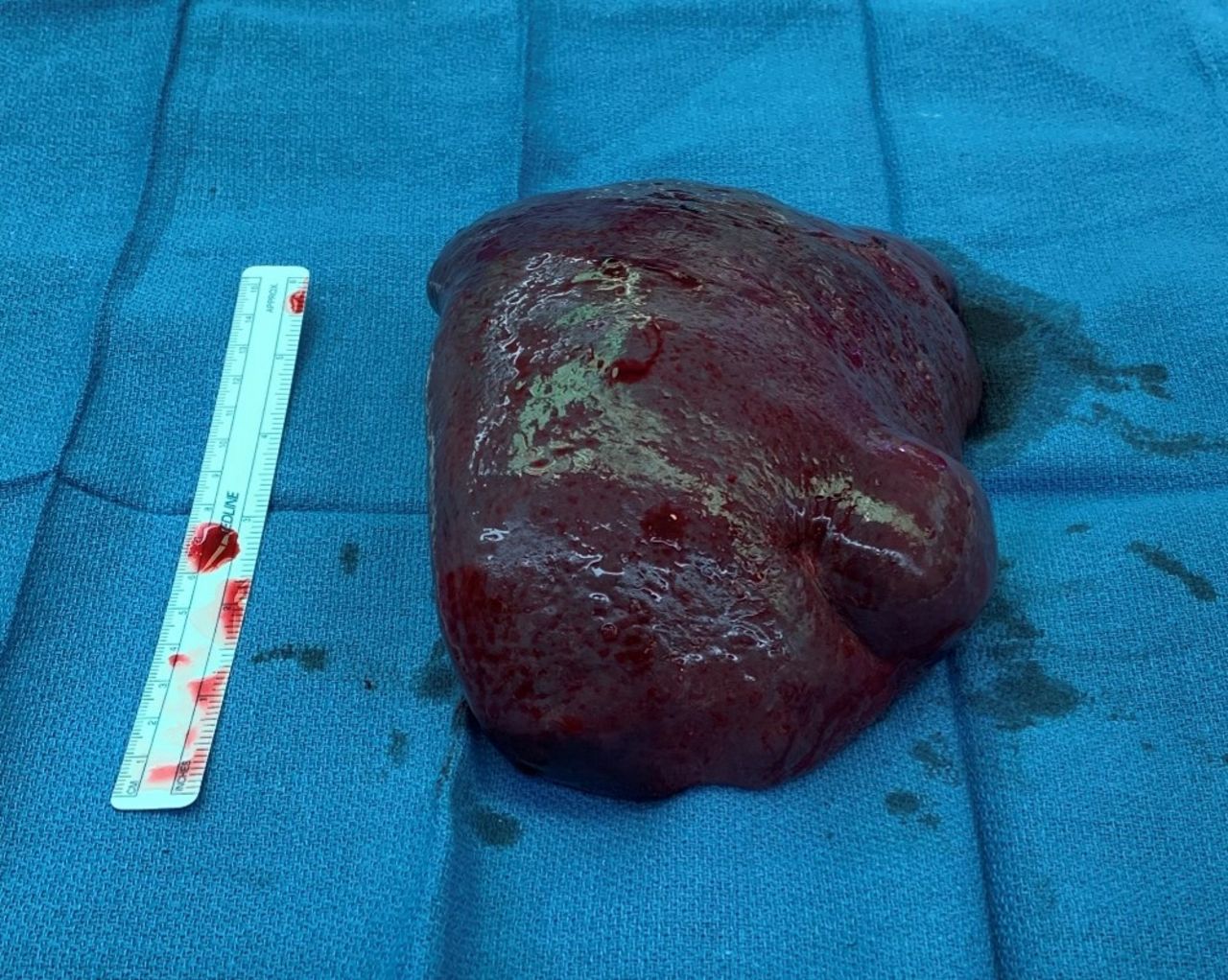
Surgical specimen after laparoscopic hand-assisted splenectomy.
The decision to operate was controversial in light of his severely deconditioned state, steroid requirement, and thrombocytopenia. His neurological function at the time of surgery could certainly be interpreted as a poor prognostic sign, suggesting palliative measures may have been more appropriate. However, given the rapidity of his decline and high-functioning status just weeks prior to his admission, we favored a potentially reversible process. Partial splenectomy is rarely warranted in an adult patient due to postoperative bleeding risks and could not have been safely performed for this lesion. The diagnostic yield of bone marrow biopsy is low in this situation and the associated delay could lead to irreversible neurological decline. Although steroid administration was initially associated with some improvement, rapid neurological decline ensued and chemotherapy was unlikely to be tolerated in his near comatose state.
He tolerated the operation well without complications. Postoperatively, he experienced swift recovery of neurological function and resolution of thrombocytopenia. On postoperative day 1, he was following commands and ambulating. By postoperative day 7, he exhibited near-complete neurological and hematological recovery. His marked recovery supported the assertion that his decline was related to a paraneoplastic syndrome from his hematological malignancy. Histopathology of the splenic mass confirmed DLBCL.
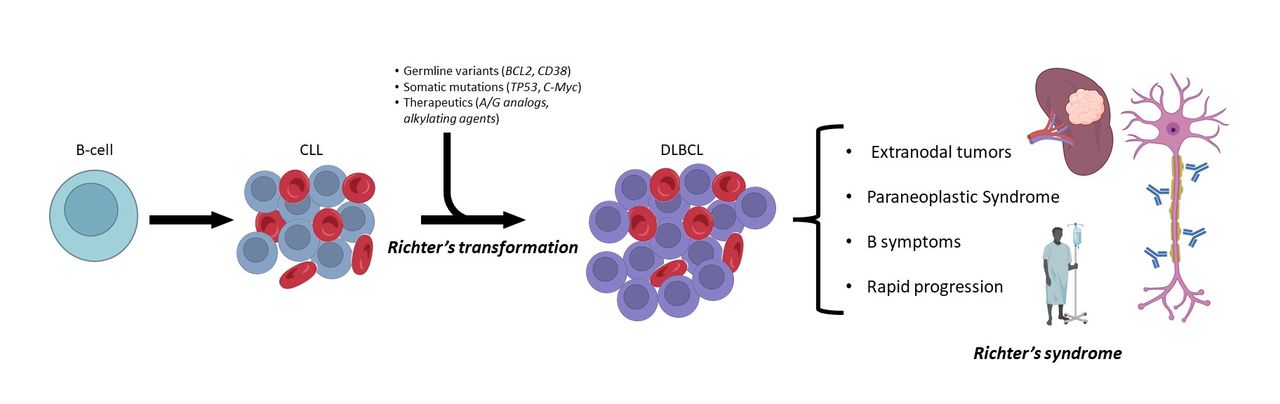
This patient course highlights two important phenomena in the progression of lymphoproliferative disorders. First, Richter’s syndrome is a broad term describing transformation of low-grade lymphoproliferative disorders to more aggressive lymphomas, the most common being CLL to DLBCL (figure 3). Richter’s transformation is multifactorial, associated with genetic aberrations, immunosuppression, and certain therapies used in the treatment of CLL, namely purine analogs and alkylating agents. When such transformation takes place, patients may rapidly decline, especially in cases of clonal expansion from the primary malignancy with early signs of disease, manifesting as lymphadenopathy, extranodal spread, or multiorgan failure. Second, the initial presentation of this patient was a rapid neurological decline—a paraneoplastic syndrome indicative of specific targeting of the central nervous system (CNS). Paraneoplastic syndromes of the CNS have previously been described in cases of aggressive lymphomas, notably DLBCL, where patients develop neuropathies and cerebellar degeneration due to overproduction of cytokines or antibodies, produced by malignant B cells, targeting CNS antigens. Paraneoplastic syndromes are exceedingly rare, with <1% incidence in solid tumors and even less frequent in lymphoma. Interestingly, the extranodal tumor burden in this patient was directly related to the severity of the paraneoplastic syndrome, with resection resulting in rapid resolution.
{kind=link}
{kind=link}
{kind=link}
Natural history of Richter’s transformation from chronic lymphocytic leukemia (CLL) to diffuse large B cell lymphoma (DLBCL).
Unless there is a mechanical complication associated with lymphoma, such as an obstruction or perforation, surgical resection is not the treatment of choice. Instead, chemotherapy is the optimal treatment for this otherwise systemic disease. However, in this case, there was a single focus of malignancy that could be resected without undue morbidity. His mental status then improved to the point that he could begin definitive treatment with chemotherapy. This case further highlights the importance of a multidisciplinary management of complex oncological cases with atypical presentations.
Ethics statements
Patient consent for publication
Footnotes
Contributors HM and DGS were responsible for literature and case review, article drafting, and article revisions. RRN was responsible for project conception, case review, and article revisions. DWB was responsible for case review and article revisions. DD was responsible for project conception, case review, and article revisions. All authors approve of the final article version.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; internally peer reviewed.