Article Text

Abstract
Background Intraosseous (IO) drug delivery may be necessary in emergency situations when intravenous access is unattainable. Valproic acid (VPA) is a histone deacetylase inhibitor that has previously been shown to improve survival in preclinical models of lethal polytrauma. In this study, we sought to compare serum levels of intravenously and IO-delivered VPA, and to analyze the effect of IO-delivered VPA.
Methods Swine were subjected to 40% blood volume hemorrhage, brain injury, femur fracture, rectus crush injury and liver laceration. After 1 hour of shock, animals were randomized (n=3/group) to receive normal saline resuscitation (control), normal saline+intravenous VPA 150 mg/kg (intravenous group) or normal saline +IO VPA 150 mg/kg (IO group). Serum levels of VPA were assessed between groups, and proteomics analyses were performed on IO and control groups on heart, lung and liver samples.
Results Intravenous and IO serum VPA levels were similar at 1, 3, 5 and 7 hours after starting the infusion (p>0.05). IO-delivered VPA induced significant proteomics changes in the heart, lung and liver, which were most pronounced in the lung. Biologic processes affected included inflammation, metabolism and transcriptional & translational machinery. The control group had 0% survival, and the intravenous and IO group both had 100% survival to the end of the experiment (p<0.05).
Discussion IO-delivered VPA is noninferior to intravenous administration and is a viable option in emergent situations when intravenous access is unattainable.
Level of evidence Not applicable (animal study).
- proteomics
- brain injuries
- traumatic
- hemorrhage
- histone deacetylase inhibitors
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Intraosseous (IO) infusion of drugs is a popular route of delivery in emergent situations. IO access can be easier to obtain for emergency drug delivery, and guidelines recommend the IO route when intravenous access is difficult or impossible to obtain.1 2 This could be especially pertinent in trauma when there are extremity injuries that limit available intravenous access sites or hemorrhagic shock associated with hypovolemic vein collapse that makes intravenous access challenging. In such situations, IO access is considered a safe alternative to intravenous access and has high success rates (90% to 100%) for achieving suitable access for emergent drug delivery.3 4 However, the pharmacokinetic (Pk) profile differs for some drugs when administered IO compared with intravenous, which would likely change dosing strategies.5
Valproic acid (VPA) is a medication that is part of the class of drugs known as histone deacetylase inhibitors (HDACI). Preclinical studies in realistic swine models of lethal hemorrhage and polytrauma have demonstrated that early administration of VPA improves survival.6–9 In survival models of traumatic brain injury (TBI), VPA decreases brain lesion size and improves neurocognitive outcomes.10 11 These studies, however, all entailed administration of VPA via a peripheral intravenous infusion. Therefore, the efficacy and pharmacokinetics of VPA-delivered IO have not been evaluated or validated.
Establishing the reliability of IO-delivered VPA is important as translation to human use for trauma will occur soon in already funded phase II/III clinical trials. In this study, we used a previously established swine model of lethal hemorrhage and polytrauma to test administration of VPA via the IO route. We hypothesized that IO-delivered VPA would be noninferior to intravenously delivered VPA in regard to peak serum concentration and area under the curve (AUC).
Methods
Animals
In conducting research using animals, the investigators adhered to the Animal Welfare Act Regulations and other Federal statutes relating to animals and experiments involving animals and the principles set forth in the current version of the Guide for Care and Use of Laboratory Animals, National Research Council. All animal protocols were approved by the University of Michigan Institutional Animal Care & Use Committee under protocol # 00007823. Female Yorkshire swine weighing 39 to 42 kg were used for the experiments to control for a gender dimorphism that could exist (Michigan State University, East Lansing, MI). After induction with Telazol (Pfizer, New York, NY). Animals were kept under general anesthesia with isoflurane for the duration of the experiment.
Injuries
The animal protocol and injury pattern has been described in detail previously, and a timeline of the experiment is outlined in figure 1.6 Briefly, animals underwent placement of femoral venous and arterial lines, a pulmonary artery catheter and open cystostomy for invasive monitoring, hemorrhage and resuscitation. For the injury protocol, animals had a rectus muscle crush injury on one side, femur fracture, and grade V liver laceration. The abdomen was then packed, and animal flipped prone in preparation for TBI and hemorrhage. A 12 mm depth TBI was inflicted using a controlled cortical impact device, and the animal was simultaneously hemorrhaged 40% of its estimated blood volume over 20 min. At the completion of hemorrhage, the animal was left unresuscitated and in shock for 1 hour to simulate medic response time.
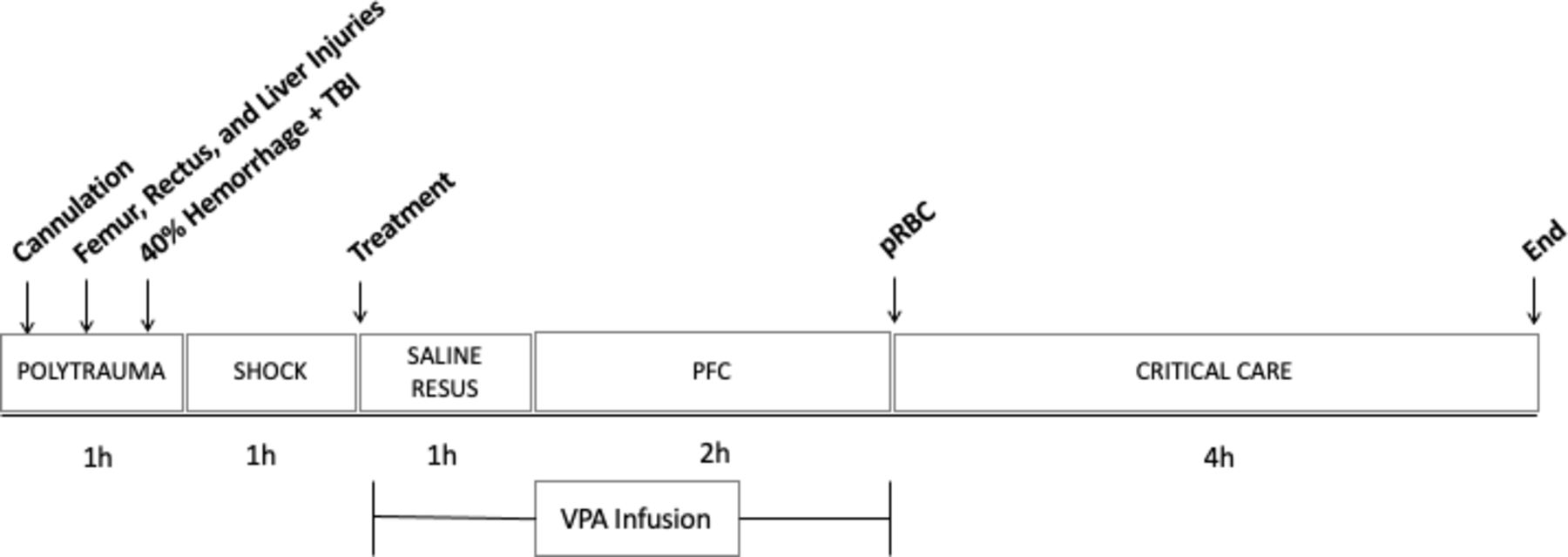
Timeline. Invasive lines placed (cannulation), followed by rectus, femur and liver injuries. Animal was then flipped prone for hemorrhage and TBI. After hemorrhage, a 1-hour shock period began, followed by normal saline resuscitation. VPA infusion was started concurrent to starting NS resuscitation and lasted 3 hours. After VPA infusion, autologous packed red blood cells were given, and then the animal was monitored for an additional 4 hours prior to end of the experiment. PFC, prolonged field care; TBI, traumatic brain injury; VPA, valproic acid.
Resuscitation and treatment
At the end of the shock phase, to simulate available resuscitative fluids available in a field care setting, all animals were resuscitated with normal saline via the femoral venous catheter with a standardized volume that was three times the hemorrhaged volume delivered over 1 hour. Animals were randomized to receive either a VPA administered IO (IO group; n=3) or administered intravenously (intravenous group; n=3) or to a control group (n=3). The primary operator and study persons caring for the animals were blinded to group allocation. VPA was administered via an IO needle placed in the left humeral head (Dieckmann Intraosseous Infusion Needle, Cook Medical, Bloomington, IN) or via a 20-gauge peripheral intravenously placed in an ear vein. Placement of the needle was confirmed by aspiration of marrow and subsequent flush of 10 mL normal saline without extravasation. A dose of VPA 150 mg/kg was administered via the IO needle or intravenously over 3 hours. To simulate a delayed transfer to definitive care, 3 hours after the start of normal saline resuscitation, animals were transfused autologous packed red blood cells that were hemorrhaged earlier in the experiment. Animals were then monitored for another 4 hours to the predetermined end point. If they survived to the end of the experiment, animals were then killed with Euthasol (sodium pentobarbital 100 mg/kg, Virbec, Fort Worth, TX). Arterial blood gases were drawn from the femoral arterial line serially throughout the experiment.
VPA monitoring
Blood samples were collected at 1, 3, 5 and 7 hours after the start of VPA infusion to measure serum VPA levels. Serum VPA levels were measured using liquid chromatography-mass spectrometry.11
Proteomics
Sample preparation
Central heart, liver and lung samples were flash frozen at the time of necropsy for control and the VPA IO groups. Each sample was homogenized in modified RIPA buffer (2% SDS, 150 mM NaCl, 50 mM Tris pH8). Protein extraction was performed by mechanical disruption using 1.6 mm stainless steel beads in a Bullet Blender (Next Advance, Troy, NY). Samples were incubated at 60°C for 30 min and clarified by centrifugation. Tissue extracts were subjected to trichloroacetic acid (TCA) precipitation.12 Washed protein pellets were solubilized in 300 µL of urea buffer (8M urea, 150 mM NaCl, 50 mM Tris pH8, 1× Roche complete protease inhibitor). Protein quantitation was performed using Qubit fluorometry (Invitrogen, Carlsbad, CA).
Protein digestion
30 µg of each sample was digested with the following protocol: (1) reduced with 15 mM dithiothreitol at 25°C for 30 min followed by alkylation with 15 mM iodoacetamide at 25°C for 45 min in the dark. (2) Digested with 2.5 µg sequencing grade trypsin (Promega) at 37°C overnight. The final digest volume was 0.5 mL adjusted with 25 mM ammonium bicarbonate. (3) Digests were cooled to room temperature and terminated with 5 µL of formic acid. The digests were centrifuged at 10 000 g for 10 min and desalted using an Empore SD solid phase extraction plate (3M, Eagan, MN). (4) Samples were lyophilized and reconstituted in 0.1% trifluoroacetic acid (TFA) for analysis.
Mass spectrometry
2 µg of each digested sample was analyzed by nano liquid chromatography - tandem mass spectrometry (LC-MS/MS) with a M-Class HPLC system (Waters, Milfrod, MA) interfaced to a ThermoFisher Fusion Lumos mass spectrometer (Waltham, MA). Peptides were loaded on a trapping column and eluted over a 75 µm analytical column at 350 nL/min; both columns were packed with Luna C18 resin (Phenomenex, Torrance, CA). A 3-hour gradient was employed. The mass spectrometer was operated in data-dependent mode, with the Orbitrap operating at 60,000 full width at half maximum (FWHM) and 15,000 FWHM for MS and MS/MS, respectively. APD was enabled and the instrument was run with a 3 s cycle for MS and MS/MS.
Data analysis
Data were searched using a local copy of Mascot (Matrix Science, Boston, MA) with the following parameters: (1) enzyme: Trypsin/P, (2) database: UniProt Pig (concatenated forward and reverse plus common contaminants), (3) fixed modification: carbamidomethyl (C), (4) variable modifications: oxidation (M), acetyl (N-term), Pyro-Glu (N-term Q), Deamidation (N/Q), (5) mass values: monoisotopic. (6) peptide mass tolerance: 10 ppm, (7) fragment mass tolerance: 0.02 Da, and (8) maximum missed cleavages: 2.
Mascot DAT files were parsed into Scaffold (Proteome Software) for validation, filtering and to create a non-redundant list per sample. Data were filtered using at 1% protein and peptide false discovery rate and requiring at least two unique peptides per protein. The full protein list with total spectral counts was exported to excel for fold-change calculations and t-tests. The fold change was calculated from the normalized spectral abundance factor (NSAF), specifically the average NSAF values for each sample category were compared. The NSAF was calculated as follows: NSAF=(SpC/MW)/Σ(SpC/MW)N, where SpC=spectral counts, MW=protein molecular weight in kDa and n=total number of proteins.13 A two-tailed, paired T-test was performed.
Gene ontology enrichment
Subsequently, gene ontology (GO) analysis was performed using the iPathway Guide (Advaita Bioinformatics, Plymouth, MI) with minimum thresholds of p<0.05 and fold change of at least 1.5 (log2 fold change 0.6). GO terms described the highest ranked biologic processes in the context of terms from the Gene Ontology Consortium database.14 For each GO term, differentially expressed (DE) proteins from the proteomics analysis were compared with the number of DE proteins expected by chance. We set a minimum of 5 DE expressed genes for each GO term. Redundant GO terms were manually removed.
Statistics
GraphPad Prism V.8 was used to analyze data (GraphPad Software, San Diego, CA). We conducted a power analysis to establish the noninferiority of serum VPA concentration administered IO compared with VPA administered intravenously; this was the primary endpoint of the study. A noninferiority limit of 40 µg/mL was chosen based on prior dose optimization studies,6 α=0.05, β=0.8 and SD=18. This power analysis suggests n=3/group would be necessary to establish noninferiority of IO compared with intravenous group. AUC was calculated for serum VPA levels, and repeated measures analysis of variance (ANOVA) was done to test differences between intravenous and IO group at various timepoints. Kaplan-Meier curves are presented for survival data and analyzed using log-rank comparisons. Other data are presented as mean±SD, and differences were tested using a one-way ANOVA with Tukey posthoc testing.
Results
Serum VPA
Total serum VPA AUC was similar between groups (figure 2). 95% CI for intravenous group was 293.5 to 355.8, and for the IO group, it was 301 to 469.7. At the 1-hour, 3-hour, 5-hour and 7-hour timepoints, there was no difference between intravenous and IO total serum VPA (p>0.57). Free serum VPA AUC 95% CI was 86.2 to 194.5 for the intravenous group and 165.3 to 275.6 for the IO group. At the 7-hour timepoint, the IO infusion group trended to have a higher free VPA concentration (p=0.09). At the 1-hour, 3-hour and 5-hour timepoints, free VPA was similar between both groups (p>0.59).

Serum VPA concentrations. Control, control group; IO, intraosseous group; IV, intravenous group; VPA, valproic acid.
Hemodynamics
Hemodynamic curves did not vary by group and followed an expected pattern with hemorrhage and subsequent resuscitation (figure 3). All animals exhibited tachycardia with hemorrhage. Mean arterial pressure, cardiac output and central venous pressure all decreased with hemorrhage and polytrauma and subsequently recovered with resuscitation.
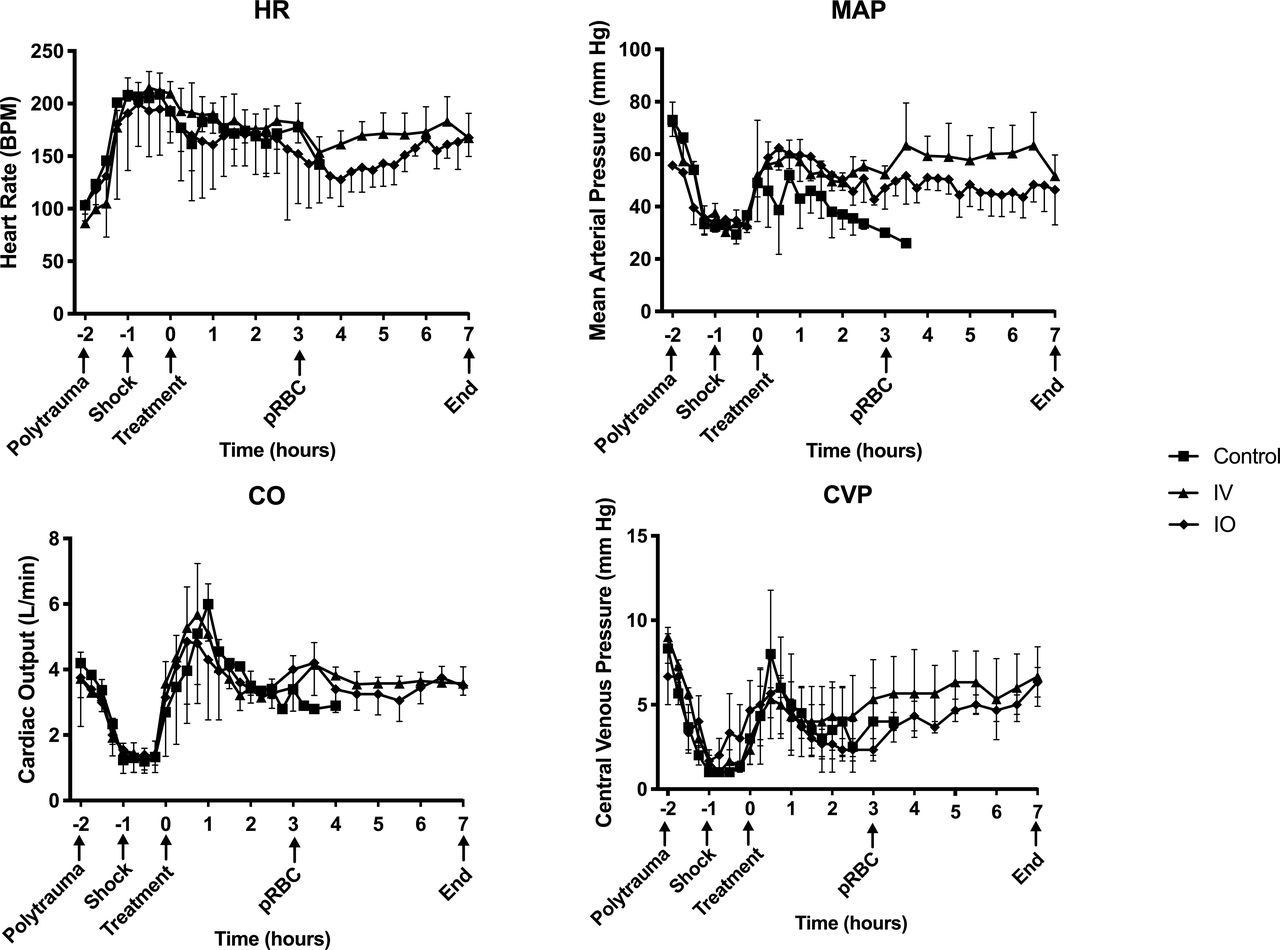
Hemodynamics curves in response to injury and resuscitation. HR measured as BPM, MAP measured in mm Hg, CO measured in liters per minute (L/min) and CVP measured in mmHg. X-axis shows hours since start of VPA infusion with important experiment timepoints denoted. BPM, beats per minute; Control, control group; CO, cardiac output; CVP, central venous pressure; HR, heart rate; IO, intraosseous group; IV, intravenous group; MAP, mean arterial pressure; VPA, valproic acid.
Arterial blood gas
Animals in all groups became acidotic after hemorrhage and injury as reflected by a decreasing pH, HCO3 and increase in lactate (online supplemental table S1). There were no significant differences between groups.
Supplemental material
Survival
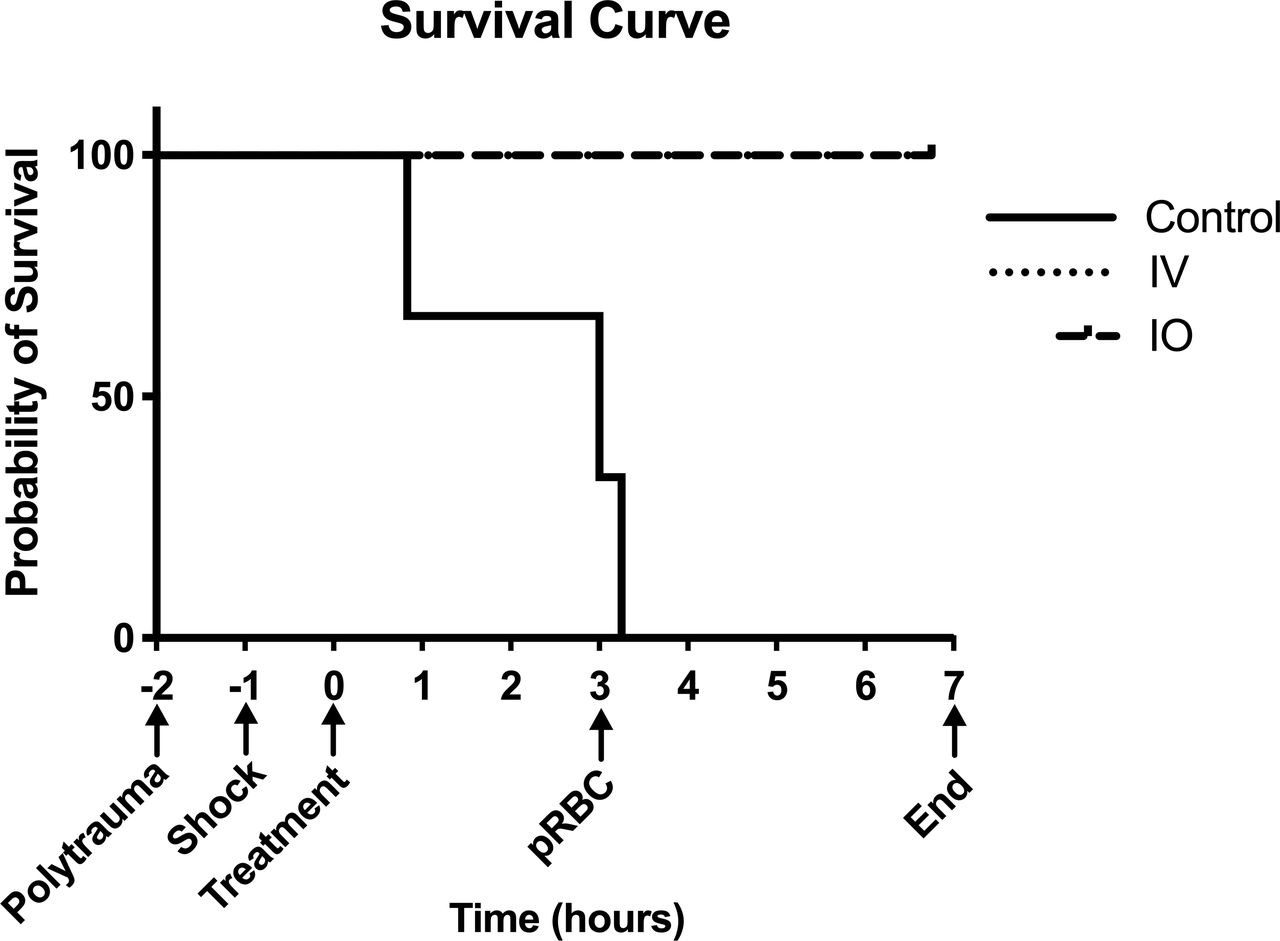
Survival is depicted in figure 4. Control animals had 0% survival, and both intravenous and IO groups had 100% survival to 9 hours after injury, at which point they were killed (p<0.01).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kaplan-Meier survival curves. X-axis shows hours since start of VPA infusion with important experiment timepoints denoted. Control, control group; IO, intraosseous group; IV, intravenous group; VPA, valproic acid.
Proteomics
In heart tissue, there were 81 proteins DE; in lung, there were 162 proteins DE; and in liver, there were 75 proteins DE. A full list of proteins is depicted in online supplemental table S2. Significantly enriched GO biologic processes are shown in table 1. The 10 most significantly enriched GO terms for lung were selected to show for easier visualization, as 189 GO terms were significantly enriched.
Supplemental material
GO biologic processes significantly enriched after valproic acid treatment delivered intraosseously
Discussion
Cardiac arrest literature of drugs in the Advanced Cardiovascular Life Support (ACLS) algorithm has compared IO and intravenous delivery. The Amiodarone, Lidocaine or Placebo in Out-of-Hospital Cardiac Arrest trial found that intravenous delivery of both drugs was superior to placebo for achieving return of spontaneous circulation (ROSC) by hospital admission and survival to hospital discharge. However, IO delivery was similar to placebo; intravenous versus IO comparisons were underpowered to detect a difference. Time to drug delivery was similar for IO and intravenous routes.15 The PARAMEDIC2 was a trial that compared intravenous and IO routes for prehospital delivery of epinephrine and found similar rates of ROSC and long-term survival.16 Observational studies have had mixed outcomes, though they tend to favor intravenous over IO for ACLS drugs.17–20 Meta-analysis of these studies favors intravenous over IO for ROSC and long-term survival after cardiac arrest.21
In controlled swine models of cardiac arrest, time to ROSC and Pk analyses is equivalent for intravenous compared with various models of sternal and humeral IO for delivery of amiodarone, epinephrine and vasopressin.22–26 However, some swine studies comparing intravenous and tibial IO access for epinephrine and vasopressin delivery showed faster time to peak drug concentration and higher drug concentration with intravenous delivery.22 27 28 Taken together, these data suggest that IO is at least a feasible option for emergent drug delivery when intravenous access is impossible or difficult to obtain. When achieving the maximum concentration in seconds is of the utmost importance (eg, epinephrine bolusing during cardiac arrest), perhaps the site of IO access is more relevant with more distal sites (tibia) inferior to proximal sites (humerus and sternum). In this study, we did not evaluate different sites of IO administration, though they were able to show comparable serum concentrations with intravenous and humerus IO VPA infusion over 3 hours. The long infusion of VPA likely makes rapid uptake (within seconds) of the drug less relevant compared with drugs used in ACLS situations. Interestingly, the pharmacokinetic profile of some drugs can change when administered IO, and a lower peak concentration for antibiotics and anticonvulsants administered as an infusion (similar to our VPA infusion).5 29–31 This is thought to be a depot effect in the IO space that causes a lower peak serum concentration, and a slower time to peak concentration.32 33 The similar serum concentrations we have demonstrated here would suggest that no dosing adjustment would need to be made with IO administration of VPA, which would be of practical concern for frontline medics.
To demonstrate a meaningful biologic effect of IO-delivered VPA, the proteomics analysis was done on selected organs thought to be critical to injury response. Significant genomic and proteomic work has been done previously examining the role of VPA in TBI.34–38 To our knowledge, no such work has been done on other injured organ systems after hemorrhagic shock and polytrauma. Similar to the TBI work, there were many proteins DE after VPA treatment in the heart, lung and liver. Others have seen a protective effect with HDAC inhibition in organ-specific ischemia reperfusion models of the heart,39–41 lung42 and liver,43 44 which could be analogous to the ischemia and reperfusion that occur with hemorrhagic shock and subsequent resuscitation. In this study, more of a pronounced change seems to occur in the lung compared with the heart and liver, with many more GO biologic processes affected. The mechanism for this is unclear, though suggesting that there could be somewhat of an organ-specific effect of VPA. Histone deacetylase complexes (HDAC) are variably distributed; specifically, class II HDACs have tissue-specific roles.45 Additionally, the cellular state and relative health versus injury of the cell is thought to dictate the response to HDACI treatment.46 These are likely reasons for the differential organ responses to VPA in this model.
Consistent with the TBI genomic and proteomic work, there were many GO biologic processes involved in the inflammatory response, cellular metabolism, and transcriptional and translational machinery affected, all of which could explain a cytoprotective effect. Specifically, network analysis using iPathway revealed a central suite of proteins in the lung critical for protein metabolism and translation that were enriched with VPA treatment including ribosomes (RPS20, RPS28, RPL13A, RPL8, RPL21, RPLP1), translation initiation (EIF4B, EIF4E) and proteasome regulation (PSMA6, PMB3, PSMB4, PSMB8, PSMD9). The most significantly affected pathway in the lung was NIK/NF-kappaB signaling. Specific proteins in our dataset that most significantly attributed to this enriched GO term include a downregulation of programmed cell death proteins, coactivators of NF-kB related transcription (ACTN4), phosphatases involved in cell stress response (PSMD9) and a host of proteasome subunits involved in ATP/ubiquitin-dependent peptide cleavage; taken together, this likely reflects a general shift away from a catabolic and cell death state towards an anabolic and cell survival state. Liver and heart network analysis differed from the lung tissue; however, the central suite of proteins was highly conserved between the heart and liver. This central suite in the network analysis for heart and liver included those involved in oxidative metabolism (ATP synthase subunits) and RNA transcription (LSM4, SNRPD1, SRRM2). The reason for this difference between lung and heart and liver is unclear though it demonstrates organ-specific effects of HDACs and warrants further investigation.
A limitation of this study is the small sample size, though a priori power analysis suggested the study was adequately powered to demonstrate noninferiority of serum VPA concentration-delivered IO compared with intravenous ones. It appears from these data that VPA confers a survival advantage in this model of polytrauma. Though we are reluctant to make strong claims on the survival benefit from VPA given the small sample size in this study, previous studies with more power have also demonstrated that VPA can improve survival in swine models of polytrauma and hemorrhagic shock.6 If a survival difference exists between intravenous and IO administration, this presented work is not adequately powered to detect that difference, and a much larger study would need to be done. Ethical and cost considerations would be prohibitive to doing such a study. However, if serum concentrations of VPA are similar between intravenous and IO administration, it stands to reason that survival benefit would be equivalent. Additionally, cost considerations limited performing the proteomics analysis on the intravenous group. However, again with similar serum concentrations of intravenous and IO VPA, we felt this analysis would not have added significant meaning to the study. Finally, in performing pilot experiments, it became clear that maintaining a continuous and accurate IO infusion of VPA was very difficult with various infusion pumps. The reason for this is unclear but could be related to the slow infusion rates and associated difficulty in maintaining IO needle patency. Hand injection with a syringe attached to the IO line easily abrogated this, though this is not practical in the clinical setting and will need to be troubleshooted in the future. Our lab is actively working on addressing this issue.
In conclusion, we have demonstrated here that IO infusion of VPA achieves similar serum levels of VPA compared with intravenous infusion and this results in a similar survival benefit. Again, we have replicated the survival benefit of a single dose of VPA given early after injury in a model of hemorrhage and polytrauma. Demonstrating noninferiority of IO VPA is an important and practical step given the difficulty of gaining intravenous access in emergent settings, especially with extremity injury and hypovolemia.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
MPP and HBA are joint senior authors.
Contributors BEB, MP and HA contributed to experimental design. BEB, ROC, MTK, GKW, AMW, MP and HA contributed to data collection and analysis and drafting of the article.
Funding This work was supported by the Frederick A. Coller Society Surgical Research Fellowship awarded to BEB. BEB was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number F32GM130010.
Disclaimer The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All animal protocols were approved by the University of Michigan Institutional Animal Care & Use Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. Proteomics dataset uploaded in supplemental table.