Article Text

Abstract
There are sparse clinical data addressing the persistence of disordered coagulation in sepsis and its role in chronic critical illness. Coagulopathy in the absence of anticoagulant therapy and/or liver disease can be highly variable in sepsis, but it tends to be prolonged in patients in the intensive care unit with a length of stay greater than 14 days. These coagulation abnormalities tend to precede multisystem organ failure and persistence of these coagulation derangements can predict 28-day mortality. The studies evaluated in this review consistently link sepsis-associated coagulopathy to poor long-term outcomes and indicate that disordered coagulation is associated with unfavorable outcomes in chronic critical illness. However, the causative mechanism and the definitive link remain unclear. Longer follow-up and more granular data will be required to fully understand coagulopathy in the context of chronic critical illness.
- sepsis
- blood coagulation
- multiple organ failure
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Sepsis is defined as a dysregulated host response to infection causing life-threatening organ dysfunction1 and is marked by both inflammatory and immunosuppressive phases with tremendous morbidity and mortality. For the surgeon, sepsis is understood as both a critically time-sensitive condition with need for immediate source-control measures, as well as a potentially chronic illness with downstream effects on morbidity and mortality for many years after diagnosis. Despite increased national and hospital-led public health campaigns to raise awareness and improve the timeliness of diagnosis, mortality from acute sepsis approaches 25% or even higher depending on its severity.2 Sepsis is a heterogeneous disease state with distinct phenotypic signatures,3 but generally speaking, sepsis survivors diverge into two categories: those who fully recover and those who enter the continuum of chronic critical illness (CCI).4
Disordered coagulation is a hallmark of sepsis, affecting 50% to 70% of patients,5 with alterations in coagulation ranging from benign laboratory aberrations to disseminated intravascular coagulopathy (DIC).6 The severity of sepsis-associated coagulopathy is directly linked to worse outcomes, including inpatient length of stay (LOS) and mortality.7 8 Coagulation abnormalities during CCI are less clearly understood. However, there is growing evidence that coagulopathy may distinguish patients at greatest risk for poor outcomes after sepsis. A recent prospective study found that 19% of patients with CCI had coagulopathy at admission compared with only 3% of sepsis ‘rapid recovery’ survivors, suggesting that coagulation abnormalities may identify patients more prone to CCI and persistent multiple organ failure (MOF).9
In this review, we will briefly outline the pathophysiology of sepsis-associated coagulopathy and DIC. We will then examine the existing literature on coagulopathy and associated outcomes in sepsis survivors and patients with CCI with the objective of highlighting the need for further dedicated research in this field.
Coagulation and sepsis
Historically, two pathways describe the initiation of coagulation (figure 1).10 The intrinsic or contact pathway begins with the activation of coagulation factor XII; the extrinsic or tissue factor (TF) pathway begins with TF release usually after endothelial injury. In the common final pathway of coagulation, activated factors V and X, calcium, and prothrombin unite to generate thrombin, which converts fibrinogen to fibrin. In turn, fibrin activates platelets, which crosslink to augment clot strength. Altogether, these pathways constitute the ‘coagulation cascade’, and form the basis of hemostasis.

Alterations to the known coagulation cascade in severe sepsis. Factors highlighted in red indicate a downregulation, and green indicates an upregulation during sepsis. These alterations have been shown to persist during chronic critical illness.
Excessive thrombosis, on the other hand, is a process in which coagulation and platelet activation impede vascular blood flow, causing tissue malperfusion, ischemia, injury, and necrosis.11 Driven by an exaggerated systemic inflammatory response, numerous physiologic alterations favor thrombosis during sepsis (figure 1).12 There is upregulation of procoagulant pathways initiated by a surge of circulating TF, which interacts with inflammatory cytokines IL-1, IL-6, and TNF-α.13 Platelet activating factor accelerates thrombus formation through increased TF secretion and platelet and leukocyte adhesion, creating an activated surface for further thrombin propagation.10 The hypercoagulability of acute sepsis is further bolstered by an acute endotheliopathy14 and dampening of native anticoagulation mechanisms, including TF pathway inhibitor, activated protein C (APC), and antithrombin (AT).6 Finally, the rise in plasminogen activator inhibitor (PAI) and formation of thrombin-activatable fibrinolysis inhibitor enhances clot firmness during acute sepsis. Altogether, the decreased native anticoagulation pathways, along with upregulation of factors that promote clot firmness, lead to an overall resistance to fibrinolysis.6
The endotheliopathy of septic shock is in part due to catecholamine-induced damage to the microvasculature, and has been shown in sepsis and in other causes of shock such as trauma and cardiac arrest. Catecholamine-induced systemic damage results in shedding of the endothelial glycocalyx layer, leading to endothelial inflammatory changes and increased capillary leakage due to failure of tight junctions, producing a procoagulant microvasculature.15 16 The breakdown of the glycocalyx barrier allows for platelet and leukocyte adhesion to endothelial cells,17 further reducing oxygen delivery because of microvascular thrombosis, which perpetuates the cycle and ultimately leads to end-organ damage and failure.18
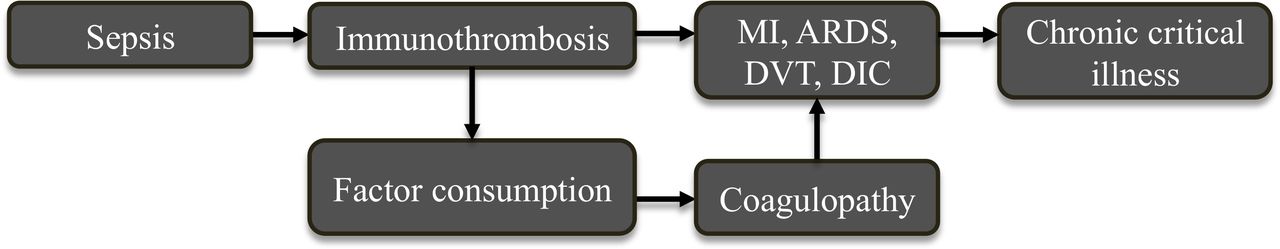
In recent years, the term immunothrombosis has emerged to more precisely describe the interplay between the host immune defense and coagulation after an infectious insult.19–21 During immunothrombosis, invading pathogens attract immune cells, which release TF and activate the extrinsic pathway of coagulation. This promotes the formation of a fibrin plug to physically barricade the microvascular circulation and protect downstream organs from further exposure to invading organisms. As the developing microthrombi continue to attract immune cells, this facilitates containment and clearance of pathogens. Although this mechanism is theoretically beneficial and evolutionarily conserved,22–25 ongoing or dysregulated thrombus formation may lead to consumption of coagulation factors, bleeding, and tissue damage, inciting pathogenic events that lead to myocardial infarction, stroke, deep venous thrombosis, acute respiratory distress syndrome, and/or DIC (figure 2).23 26–28

{kind=link}
{kind=link}
Immunothrombosis, a known phenomenon during sepsis, leads to an increase in thrombotic events, precipitates factor consumption, and propagates coagulopathy in the progression to chronic critical illness. ARDS, acute respiratory distress syndrome; DIC, disseminated intravascular coagulopathy; DVT, deep venous thrombosis; MI, myocardial infarction.
DIC is the most devastating manifestation of sepsis-associated coagulopathy, as it involves concomitant coagulation and bleeding, and impacts an estimated 13% to 61% of septic patients depending on the scoring system referenced.29–34 The International Society on Thrombosis and Hemostasis (ISTH) defines DIC as ‘an acquired syndrome characterized by the intravascular activation of coagulation with loss of localization arising from different causes. It can originate from and cause damage to the microvasculature, which if sufficiently severe, can produce organ dysfunction’.35 DIC results from activation of TF, insufficient control of coagulation, and elevated PAI levels, leading to altered fibrinolysis.28 36 Although there are numerous other etiologies of DIC including trauma and pancreatitis, in the context of sepsis, DIC may represent excessive immunothrombosis, leading to pervasive microthrombotic disease as the host attempts to contain invading pathogens systemically.26 This widespread formation of microvascular thrombi causes cellular apoptosis and necrosis with release of proinflammatory intracellular proteins, leading to multisystem organ dysfunction. In addition, the concomitant consumption of coagulation factors can cause even further issues with coagulopathy, including bleeding complications.
While DIC is considered an independent predictor of mortality during critical illness and is associated with a 1.5-fold greater mortality rate in septic patients compared with those with less severe forms of coagulopathy,36 37 no single test is sufficient for detecting DIC. In the appropriate clinical setting, accurate diagnosis requires examining numerous laboratory tests, including platelet count, fibrin markers, fibrinogen, and prothrombin time (PT). Because of the simultaneous activation and downregulation of separate parts of the coagulation process during sepsis, timely monitoring of coagulation changes is important in the recognition and treatment of sepsis-induced coagulopathy and DIC. In current practice, thromboelastography (TEG) is the most efficient and advantageous measure of clotting function in real time.38 Specific indicators in TEG testing, such as R value, K time, α angle, maximum amplitude value and coagulation index, have been shown to correlate with the severity of the patient’s sepsis by both Sequential Organ Failure Assessment and Acute Physiology and Chronic Health Evaluation II scores, and can better determine platelet functionality than traditional coagulation laboratories.39 Many studies have been conducted to evaluate the utility of TEG on clinical diagnosis of sepsis-induced coagulopathy.40–43 Among these, wide variation is seen with regard to detection of hypercoagulability and hypocoagulability, likely from non-uniform collection times, varying degrees of disease severity, and deviations in definitions.44 However, overall trends indicate that a hypocoagulable profile is associated with increased MOF, along with increased mortality in sepsis.45
Multiple scoring systems have attempted to standardize the diagnosis of DIC, but each has respective drawbacks, including broad applicability and questionable sensitivity. The most recent accepted diagnostic criteria are the ISTH overt DIC score, based on platelet counts, fibrin-related markers (D-dimer, and so on), PT and fibrinogen levels. This has been criticized for its detection of DIC in its later phases, and other guidelines have been proposed to diagnose DIC earlier in the disease course to better facilitate treatment, namely Japanese Association for Acute Medicine DIC score, and Japanese Society on Thrombosis and Hemostasis DIC.46
Unfortunately, there are no current therapeutic interventions guaranteed to reverse DIC and the mainstay of treatment depends on resolving the underlying pathophysiologic process. Because coagulopathy and microthrombotic disease are believed to contribute to organ dysfunction during sepsis, numerous clinical trials have investigated whether coagulation mediators could be effective therapeutic targets. Notable mentions include the Protein C Worldwide Evaluation in Severe Sepsis (PROWESS) group, evaluating APC. APC is an endogenous protein that has both antithrombotic and profibrinolytic properties, and originally showed promise with a significant mortality reduction, particularly in patients with DIC.47 Other studies showed APC reduced overt thrombotic events, although with an increase in clinically significant bleeding events.31 After increasing doubt in the efficacy of APC, and higher concerns for bleeding, an investigation into the risk/benefit profile of the drug was called for. The study, PROWESS-SHOCK, eventually proved no difference in 28-day survival and the drug was discontinued.48
In a similar vein, antithrombin supplementation has been studied to improve outcomes in patients with sepsis-induced DIC. Antithrombin in an endogenous anticoagulant, inhibiting 80% of coagulation activities of thrombin and five different coagulation factors. High-dose supplementation has shown possible improved survival in patients with sepsis-induced DIC, but no survival differences in patients with sepsis without DIC.49 However, more recent studies have shown that specific patient populations with sepsis-induced DIC and very low antithrombin levels (<43%) benefit from administration of antithrombin (AT), with significantly improved survival and without increased bleeding complications.50
Interpretation of these clinical trials is complicated because the benefits of ameliorating coagulopathy must be balanced against the risk of weakening the host response to infection or causing hemorrhage. Furthermore, because sepsis is heterogeneous and the pathogenesis of coagulopathy is multifactorial, simultaneous treatments and/or a personalized algorithm may be needed. These treatment modalities may be more successful once a more complete picture of the progression of coagulopathy and DIC in critical illness is available. It is a fluctuating state of imbalance between procoagulation and antithrombosis and, to accurately treat a patient, we will need to better predict their coagulation state in real time. The use of a procoagulant or anticoagulant is more likely to harm the patient if used in a manner not tailored to the specific coagulopathy in real time. One potentially viable approach is to target the innate immune cells—namely neutrophils and monocytes—or cellular products that mediate immunothrombosis to mitigate progression to DIC and/or MOF. For example, neutrophil extracellular traps (NETs), chromatin-based antimicrobial structures released from neutrophils, are implicated in propagating immunothrombosis. In preclinical models and in vitro, inhibition of NET formation may be a promising option for therapeutic intervention.51–53
Coagulation and CCI
With increased efforts to expeditiously diagnose and treat sepsis, short-term outcomes are improving significantly and the overall incidence continues to rise. Although in-hospital mortality has declined, up to 40% of septic patients fail to fully recover and progress to CCI, defined as an intensive care unit (ICU) LOS of greater than 14 days with evidence of organ dysfunction.4 54 55 Patients with CCI are plagued by recurrent infections, malnutrition, cognitive impairments, poor rehabilitation, and indolent death. They necessitate higher resource utilization and suffer significantly worse health-related quality of life and functional outcomes as soon as 3 months after their initial sepsis diagnosis.9 Chronically ill sepsis survivors have estimated 1 and 3-year mortality rates of 55% and 71%, respectively, many of which result from withdrawal of care.9 55–57 Longer term outcomes and survival data are likely to be under-reported.
In response to this newly emerging patient population of sepsis survivors, critical illness paradigms have evolved dramatically in recent years. Persistent inflammation, immunosuppression, and catabolism syndrome (PICS) was recently proposed as a mechanism to explain the subset of patients with CCI who fail to re-establish immunologic and metabolic homeostasis after resolution of their acute insult—usually sepsis, trauma, or pancreatitis.58 Physiologically, PICS involves increased circulating myeloid cells, lymphocyte suppression, and muscle catabolism. There are no consensus clinical guidelines for diagnosis of PICS, but some have characterized it as follows: ICU LOS >14 days, C-reactive protein 150 µg/dL, total lymphocyte count <0.80×109/L, >10% weight loss during hospitalization or body mass index <18, creatinine height index <80%, serum albumin <3.0 g/dL, prealbumin <10 mg/dL, and retinol binding protein <10 µg/dL.58–60 Unfortunately, beyond supportive care and antibiotic therapy, there are no life-quality or life-prolonging interventions for PICS at the present.
Few studies have assessed coagulation in preclinical models of late sepsis or CCI. Our laboratory found that outbred mice were hypercoagulable 2 hours after sepsis in a process driven by macrophage production of TNF-α.61 However, 16 hours after septic injury, mice shifted to a hypocoagulable phenotype, suggesting that early thrombosis led to coagulation factor consumption. In follow-up work, we evaluated whether dysregulated coagulation was present at even later time points after sepsis.62 To develop this murine model, mice were subject to a moderate-severity cecal ligation and puncture injury and killed 8 days later when they concurrently developed all the characteristics of PICS, as previously described.63 These PICS mice demonstrated increased splenic megakaryocyte accumulation without a concomitant increase in circulating platelets, as well as significant pulmonary microvascular thrombi with corresponding changes in respiratory function. Because dysregulated coagulation was associated with development of tissue damage and end-organ dysfunction, we concluded that coagulopathy should be considered an additional feature of PICS.
There are very little clinical data addressing the persistence of disordered coagulation, let alone its prognostic or predictive implications, during CCI. In one systematic review from Guirgis et al, persistent organ dysfunction was evaluated in survivors of severe sepsis.64 The authors included patients from five randomized controlled trials only if follow-up exceeded 28 days and if objective assessment of both organ function and resolution was performed. None of the studies evaluated organ dysfunction greater than 30 days after sepsis admission. DIC was diagnosed in 23% to 88% of patients across three of the studies. Rates of resolution for DIC were also highly variable, ranging from 12% to 87% in the three qualifying studies. Limitations included the small sample size and the reliance on different definitions of DIC. Only one study enrolled subjects during the modern era of goal-directed therapy, making it difficult to draw applicable conclusions about critical illness and coagulation.
In a subsequent study from Guirgis et al, 110 septic patients were prospectively screened for the development of persistent (28–90 days) or long-term (greater than 90 days) organ dysfunction.65 Coagulopathy was defined as an international normalized ratio (INR) >1.5 and a platelet count <100 x 10∧9/L in the absence of anticoagulant therapy and/or liver disease. The overall rate of coagulopathy at admission was 25%, consistent with the previously discussed study.9 Among sepsis survivors with available data, 10% had persistent coagulopathy and 3% had long-term coagulopathy. Clinical outcomes of patients with coagulopathy were not evaluated, likely because the sample was underpowered to establish statistical trends. Interestingly, the authors found a significant association between elevated INR and persistent organ dysfunction, but they did not perform a multivariable analysis to determine whether coagulopathy independently predicted later organ failure. Nevertheless, the concept that an abnormal laboratory test of coagulation could predict eventual MOF is promising for identifying and staging patients, as well as pinpointing a window of intervention to prevent sepsis-related morbidity and mortality.
A handful of studies have investigated short and intermediate-term outcomes of early coagulopathy among sepsis survivors. In one single-center prospective study of septic patients, Koyama et al found that higher plasma thrombin-antithrombin complex (TAT) and PAI-1 levels 2 days after ICU admission predicted 28-day mortality.66 In a larger multi-institution study, Dhainaut et al assessed coagulopathy markers early in patients with severe sepsis as well as 28-day mortality rates and progression to MOF.24 PT, D-dimer, antithrombin activity, protein C activity, and platelet counts were trended temporally. The majority of patients had baseline coagulation abnormalities that preceded organ failure and changes in PT, antithrombin, D-dimer, and composite coagulation parameters from baseline to the first day after severe sepsis diagnosis predicted 28-day mortality. Worsening coagulopathy was also associated with MOF development, whereas improvements in coagulation were associated with resolution of MOF. The authors concluded that coagulation is a key mediator of sepsis outcomes. Finally, Massion et al found that deficient thrombin generation 3 days after the onset of septic shock was associated with greater inpatient and 90-day mortality.42 Taken together, these studies highlight that common coagulation parameters may help risk stratify septic patients and identify who is at risk to progress to MOF or early death.
Two European studies have advanced our understanding of long-term outcomes of infection and CCI. In a multicenter cohort study of hospitalized patients who survived community-acquired pneumonia, Yende et al determined that after adjusting for age, sex, race, and comorbidities, higher levels of TAT, PAI-1, and D-dimer within 96 hours of discharge were associated with greater all-cause 1-year mortality.67 This is consistent with the previously highlighted study showing that elevated TAT and PAI-1 levels at the time of sepsis diagnosis predict 28-day mortality.66 Even after adjusting for IL-6 levels, elevated TAT and D-dimer remained significantly associated with 1-year mortality, of which the most common cause was cardiovascular disease likely from destabilization of existing atherosclerotic plaques, cardiomyocyte depression,68 69 and/or plaque progression and rupture.22 70 71 Although these patients did not technically have sepsis, these findings illustrate that, even without a dysregulated systemic immune response, inflammation and coagulopathy can persist after the primary infection has resolved. Prospective observational data from Gayat et al echo these findings, demonstrating that markers of cardiovascular ischemia at the time of ICU discharge discriminate 1-year survivors from non-survivors.72 The authors also found that a platelet count <100 000/µL at discharge was associated with a twofold increased risk of death 1 year after ICU admission. However, only one-fifth of included patients had septic shock as the reason of their ICU admission and additional coagulation markers were not specifically evaluated in this study. Together, these data suggest that persistence of abnormal hematologic markers at discharge is a warning sign and possible method for identifying patients vulnerable to later end-organ dysfunction and death. Further longitudinal research is needed to determine which particular serum markers, measurements, or trends predict worse outcomes for sepsis survivors. A summary of studies on coagulopathy among sepsis survivors is outlined in table 1.
Review of the literature regarding coagulopathy and outcomes among sepsis survivors
Although these studies establish that disordered coagulation is associated with unfavorable outcomes for sepsis survivors, there are some notable limitations stemming from non-uniform methodology. Median ICU LOS qualified only some subjects as true patients with CCI. Therefore, without subgroup analyses of mortality among patients with ICU admission >14 days, these data are less generalizable to CCI. There was also immense variability in how coagulopathy was defined, and which coagulation parameters were assessed. Although the data consistently linked sepsis-associated coagulopathy to poor outcomes, it was unclear whether the causative mechanism was deficient coagulation or poor fibrinolysis. It is also questionable whether short-term outcomes are even pertinent for CCI, which implies a more indolent course. Some argue that CCI mortality plateaus at 3 months and that longer follow-up is resource intensive and confounds survival data since patients may die to non-CCI-related causes such as cancer or trauma.9 73 While dysregulated coagulation is often observed on the day of sepsis diagnosis,7 24 42 66 longer follow-up and more granular data are required to understand coagulopathy in the context of CCI. This may be achieved through greater multidisciplinary and/or multi-institutional collaboration to foster creation of consensus CCI guidelines and standardize language for research and clinical purposes.
Conclusions
Sepsis-associated coagulopathy is a frequent and well-described clinical phenomenon that can lead to organ dysfunction. By contrast, coagulation during CCI is poorly understood with few animal or clinical studies revealing its pathophysiology, incidence, presentation, or clinical outcomes. Septic patients who experience coagulopathy are more likely to progress to organ failure, and those with improvements in coagulation demonstrate greater incidence of MOF resolution. Additionally, this disordered coagulation introduces a greater risk of morbidity during surgical source control, which has the potential to lead to an even more complicated course. However, the coagulopathy of sepsis is just one facet that leads to persistent organ dysfunction and primes patients for the development of CCI. The association between them has been proven, but the link is still unknown.
This review illustrates that coagulopathy does persist in sepsis survivors and that abnormal coagulation at the time of admission and discharge predicts mortality outcomes up to 1 year after sepsis diagnosis. There is very compelling evidence of the impact of coagulopathy on outcomes in CCI, but further studies are needed to prove more than correlation between the two. None of the discussed studies explored the association between coagulopathy, persistent organ dysfunction, and mortality among a strict CCI population. A thorough examination of the temporal trends of coagulopathy among sepsis survivors is needed to elucidate whether such abnormalities in CCI represent progression versus de novo disease and whether they directly contribute to MOF. Future clinical work should also track outcomes for a greater duration of time given that CCI incurs long-term immunologic, functional, nutritional, and lifestyle morbidity that shorter term measures fail to capture. Together, these data may help classify patients into CCI phenotypes, similar to acute sepsis.3 7 Predicting which patients with coagulopathy will ultimately develop persistent MOF and CCI is clinically useful because it may guide resource utilization decisions, facilitate family discussions about goals of care, and to some extent direct patient management in both the acute stages of disease and in the chronic illness state that follows. Unfortunately, until there are better clinical targets, treatment options, and biomarkers to follow therapeutic efficacy, the ability to minimize excessive inflammation, restore immunologic homeostasis, and prevent organ dysfunction in sepsis survivors is not yet possible. However, along with the foundations in the literature reviewed here, perhaps future studies focusing on coagulopathy in CCI may help inform our decision-making for sepsis survivors to improve overall outcomes for these patients.
References
Footnotes
LKW and CS contributed equally.
Contributors LKW and VN conceived the presented ideas. LKW and CS wrote the article with support from NB, CCC and VN. All authors provided critical feedback and helped shape the research, analysis and article.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.