Article Text

Abstract
High-quality clinical trials are needed to advance the care of injured patients. Traditional randomized clinical trials in trauma have challenges in generating new knowledge due to many issues, including logistical difficulties performing individual randomization, unclear pretrial estimates of treatment effect leading to often unpowered studies, and difficulty assessing the generalizability of an intervention given the heterogeneity of both patients and trauma centers. In this review, we discuss alternative clinical trial designs that can address some of these difficulties. These include pragmatic trials, cluster randomization, cluster randomized stepped wedge designs, factorial trials, and adaptive designs. Additionally, we discuss how Bayesian methods of inference may provide more knowledge to trauma and acute care surgeons compared with traditional, frequentist methods.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Injury is the leading cause of death and disability for those aged 1–45 and remains a significant source of morbidity and mortality for older age groups. Although there have been many advances in the care of the injured patient during the last few decades, these advances have largely been due to either technological advances in fields outside of surgery or the rapid need for knowledge generation in the treatment of casualties during war. These include angioembolization of solid organ injuries, the maturation of CT scanning, and balanced blood product resuscitation.1–6
While these advances have contributed to a reduction in mortality for some types of injured patients, others have seen no change in decades.7 This failure to improve mortality in some types of injured patients is partially due to the lack of well-designed clinical trials to guide care.8 9 Many of the best available guidelines used to inform treatment decisions are based on retrospective studies and low-quality data. To change practice and to improve morbidity and mortality for injured patients there is an urgent need for clinical trials in this space.
There are multiple barriers to performing high-quality clinical trials in injured patient populations. These include a lack of funding to allow for adequately powered studies, difficulty assessing the effect of an intervention in a heterogeneous patient population, and difficulty assessing the generalizability of interventions given significant differences between trauma centers. Additional barriers include issues with consent, difficulty in choosing appropriate outcome measures, a lack of long-term follow-up, and other factors that will not be covered in this review. In this review, we will focus on clinical trial designs other than the traditional parallel group randomized controlled trial (RCT). Alternative trial designs can be used to generate new, generalizable knowledge in an efficient manner and improve the care of injured patients. These include pragmatic, cluster randomized, cluster randomized stepped wedge, factorial, and adaptive designs. Additionally, we will contrast traditional frequentist statistical analysis with Bayesian methods of inference.
Pragmatic RCTs
An explanatory or efficacy trial is one performed with experienced investigators at advanced sites following strict enrollment criteria that ultimately creates a narrow patient population receiving the intervention under ideal circumstances. Explanatory trials are often appropriately used to test the efficacy of a new intervention or to determine the mechanistic causes for the observed treatment effect. They focus more on the internal validity of the study rather than the generalizability of the intervention. Because of the focus on internal validity at the expense of the generalizability of the study, the treatment effect that results from an explanatory trial will likely overestimate the benefits of the intervention when applied to a ‘real world’ population with more heterogeneous patients. The result is the most common question physicians ask after reading a clinical trial: Does this apply to my patients?10
Contrary to explanatory trials, pragmatic randomized clinical trials measure the effectiveness of an intervention: How does this intervention work in real clinical practice? There are typically few to no exclusion criteria in the population of interest and the intervention is provided along with routine care by a heterogeneous group of clinicians. External validity is explicitly valued during the design phase of the study to maximize the generalizability of the study.
While there is no clear line between an explanatory and pragmatic RCT, there exists a continuum on which domains of an individual trial may have varying levels of each. One scoring system used to measure a study’s pragmatism is called the PRagmatic-Explanatory Continuum Indicator Summary (PRECIS-2).11 PRECIS-2 rates an RCT on a Likert scale of 1–5 (1=very explanatory; 5=very pragmatic) in nine domains: eligibility, recruitment, setting, organization, flexibility: delivery, flexibility: adherence, follow-up, primary outcome, and primary analysis (table 1).
Domains of the PRECIS-2 tool11
The Pragmatic Randomized Optimal Platelet and Plasma Ratios trial was an example of a pragmatic clinical trial in trauma.1 In this study, patients were individually randomized to one of two balanced resuscitation strategies—1:1:1 or 2:1:1 of red cells, fresh frozen plasma, and platelets, respectively—for the primary outcome mortality. All other care at each of the 10 participating trauma centers was given as it normally would at that center, including the volume of crystalloid given during resuscitation, the utilization of hemostatic adjuncts (tranexamic acid, factor VII, and so on), or various clinical management algorithms.
In any trial, the compromises needed to make the study a reality create trials that span the explanatory-pragmatic continuum. It is vitally important that the location on this continuum be consistent with the question the investigator is asking and the conclusions that the investigator wishes to impart on the eventual reader (table 2).12 Depending on the question being asked and the conclusion desired, the time to determine pragmatism is during the design phase as a clinical trial with no consideration for external validity might result in a conclusion that has no relevance outside the patients recruited. Pragmatic trials avoid this outcome by testing the intervention in an environment that most closely resembles how it will be used in the real world (table 3).
Outcome of explanatory and pragmatic trials12
Pragmatic clinical trials
Cluster randomization
In a traditional parallel group clinical trial, patients are individually randomized to a treatment arm and the treatment arm to which that patient is enrolled is independent of other patients’ treatment allocation. When testing a new method or process of care (eg, a new care bundle), however, randomization at the individual level can be infeasible. The bundle may be incompletely implemented in the experimental arm and contamination of the control group with all or some of the intervention can occur. Both reduce treatment differentiation between the experimental and the control arm resulting in an imprecise estimate of treatment effect. Cluster randomization can address these problematic limitations of randomization at the individual level. In a cluster randomized trial, all patients in a given cluster are randomized to the same treatment arm.13 Clusters can be at any level—trauma center units, trauma centers, cities, states, regions, and so on.
The Prehospital Air Medical Plasma trial was a multicenter cluster randomized in which participating air medical bases were randomized to provide prehospital thawed plasma as the primary resuscitative fluid or usual care for patients in hemorrhagic shock.14 Cluster randomization was chosen in this trial given logistical challenges of having universal donor thawed plasma available at all of the sites, as opposed to just half of them.
While appealing when individual randomization is problematic, cluster randomization introduces some issues when interpreting the results of a trial. First, patients within clusters tend to be more similar to each other than to patients in other clusters. This is quite different compared with traditional clinical trials where patients are assumed to be independent of each other. Statistical methods that account for patient similarities within cluster are needed when analyzing the results of a cluster randomized trial, such as hierarchical or mixed effects models that control for between and within-group differences while testing the treatment effect. Second, differences in outcome due to differences in centers, as opposed to the intervention, can be difficult to accurately measure. Third, blinding is often impractical in cluster randomized trials, so efforts to ensure objectivity in measuring outcomes are even more critical than in traditional trials. Due to some of these issues, more patients are needed in cluster randomized trials compared with individual randomization to maintain the same statistical power (table 4).
Cluster randomized trials
Cluster randomized stepped wedge designs
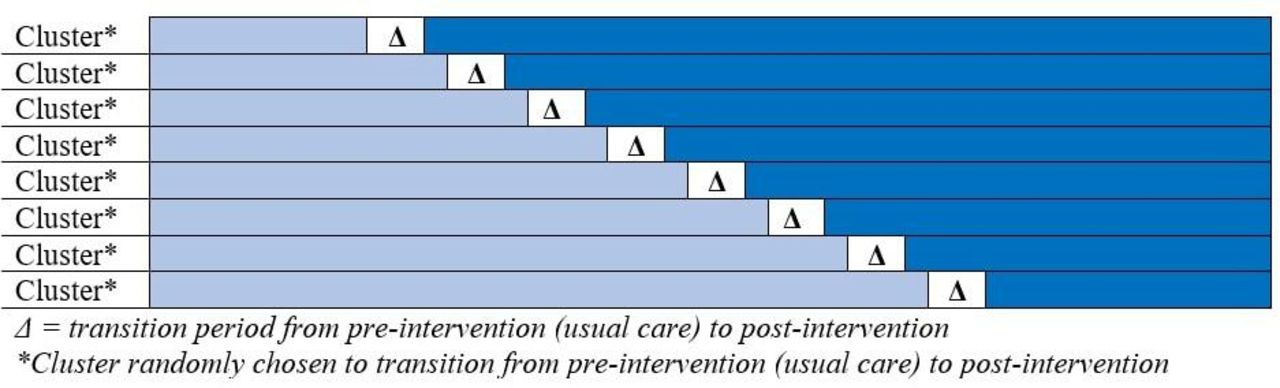
In a cluster randomized trial, clusters are randomly assigned to either a treatment or a control and followed for the entirety of the study. Then, comparisons between the treated and untreated clusters are performed once the trial is complete. A cluster randomized stepped wedge design, however, begins by following a cluster of patients prior to receiving the intervention of interest. These observations create the control group. The intervention is then applied to each cluster at a randomly selected point in time such that the cluster crosses over from the control to the intervention. Data collection continues such that each cluster eventually is exposed to the intervention. Thus, each cluster contributes observations during the control and intervention periods (figure 1).

Schematic showing the transition of clusters from usual care to intervention. In a cluster randomized stepped wedge study, all clusters begin the trial providing usual care (preintervention). After a predetermined amount of time in the preintervention period, clusters begin randomly transitioning to the intervention arm. Each cluster provides observations for both the control group (usual care) and the intervention group.
The design is commonly used in the evaluation of processes and models of care delivery in which individual randomization is problematic (table 4). The earliest and most widely known cluster randomized stepped wedge design study was the Gambia Hepatitis Intervention Study.15 This study randomly selected geographic areas in Gambia every 12 weeks in which to roll out routine hepatitis B virus (HBV) vaccination with the goal of introducing HBV vaccination to the entire country in a 4-year period. This creates a group of HBV-vaccinated and a group of HBV-unvaccinated children during that 4-year period to compare long-term outcomes contemporaneously while also allowing all geographic areas to eventually cross-over into routine HBV vaccination. Long-term data collection on the effectiveness of the vaccine in preventing hepatocellular carcinoma and other chronic liver disease is ongoing. In this example, the advantage of a cluster randomized stepped wedge design is that it allows for an intervention considered to have short-term benefits to be deployed across a wider scale to improve the health of all patients while also allowing for more rigorous evaluation of the effectiveness of the program during deployment (table 5).
Cluster randomized stepped wedge trial design
When designing a cluster randomized stepped wedge trial, the number of clusters, the length of the control and intervention periods, and the number of clusters which cross-over at any given time are typically determined by logistical considerations related to the implementation of the intervention.16 Since individuals within clusters tend to be more similar than individuals in other clusters, a larger sample size is needed relative to a traditional RCT to maintain the same power. Indeed, the adequate powering of a cluster randomized stepped wedge is the degree of variation due to within-cluster or between-cluster differences, also called the intraclass correlation. Additionally, temporal trends in the outcome of interest must be accounted for to assess for confounding due to time period.
Adaptive designs
Traditional parallel group clinical trials involve fixed randomization allocation ratios (eg, 1:1:1, and so on) that are maintained throughout the study period. A set of stopping rules enforced by predetermined thresholds or Data Safety Monitoring Boards offers the lone opportunity to evaluate the need for protocol changes during the trial. Adaptation refers to the sequential rebalancing of allocation ratios based on a predetermined response algorithm which incorporates key measures collected during the course of the trial (often the primary or secondary outcome measures).17 It uses Bayesian statistical methods of inference from prior probability distributions and the available observed data to inform a posterior probability distribution. This posterior distribution is used to adjust the allocation ratio of the trial. The adaptive trial begins at a set allocation ratio with interim analyses at planned enrollment intervals, commonly after a longer duration ‘run-in’ phase. At each interim point thereafter, the response measures of interest are incorporated and used to adjust allocation to reflect the best performing arm(s).
Adaptive designs are particularly useful when there are ambiguous or multiple comparison groups to consider and the need to ‘dial in’ the right comparison groups from many to few. The best example of this condition would be an outcome variable where the investigators have little evidence about what interventions to test in their study. Adaptive designs can allow for a range of interventions to be tested and the outcome assessed at interim analyses. Poor performing interventions (harmful or no effect) can be dropped from the study such that future patients would be allocated to other promising interventions to improve the sample size within that intervention.
One method of adaptation is response-adaptive randomization (RAR). RAR is often used in dose-finding trials, which begin with multiple different doses of a new medication meant to treat some condition.18 At interim analyses, dose levels with favorable outcomes have the future allocation ratio increased and dose levels with deleterious side effects may have the future allocation ratio decreased. An example of an adaptive design with RAR is the Stroke Prevention in Atrial Fibrillation trial.19 In this trial, patients will be randomized to one of four different treatment strategies for anticoagulation after mild, moderate, and severe stroke. At prespecified interim analyses, the allocation ratio will be altered to increase the sample randomized to the arms showing a more favorable risk-benefit profile. A second method of adaptation is sample size reassessment in which the final target sample size is increased or decreased based on the treatment effect seen at interim analyses. Many trauma and emergency surgery trials attempt to test an intervention with an unknown treatment effect resulting in an incorrect sample size calculation. Adaptive designs with sample size reassessment can ensure that, at the end of the trial, an adequate sample has been recruited to accurately estimate the effect of an intervention.20 21
There are many benefits from the use of adaptive designs, most intuitively the safety of the trial subjects (table 6). The allocation ratios adapt to assign marginally increasing numbers of the sample into the best performing arm(s). If that arm eventually begins to underperform, then allocation will rebalance accordingly. Overall, this has the effect of introducing a larger proportion of the sample to the most effective treatment, assuming one can be identified during interim analyses. This is especially beneficial in the fields of trauma or emergency medicine, where providing care based on the best available evidence can save lives and improve chances of recovery. Additionally, trials conducted with adaptive designs can achieve findings similar to frequentist trials with greater power and/or a smaller sample by improving the precision of estimates in the critical arms. The outcome test will produce a different set of information for interpretation, but the overall efficiency of the trial to detect multiple differences across arms is often improved through adaptive design.
Adaptive trial designs
Factorial designs
In the standard RCT, one intervention is assessed while maintaining similar processes of care in the control group. Randomization at the subject level balances known and unknown confounders resulting in two comparison groups for whom the only difference is the intervention. Sometimes, however, investigators want to study more than one intervention. In the setting of multiple interventions, one could perform multiple concurrent trials, perform multiple trials in succession using the winner of the previous trial compared with the next intervention, perform a parallel ≥2 group trial, or perform a trial using a factorial design. Assuming the multiple interventions work independently, the factorial trial is the most efficient manner to study more than one intervention within a single trial.22
For example, if two interventions were being assessed for a binary outcome (present or absent), there would be four treatment groups in the trial being tested—group 1 receives neither intervention, group 2 receives both interventions, group 3 receives intervention A but not intervention B, and group 4 receives intervention B but not intervention A. In this example, the effect of intervention A is determined by comparing all patients who received intervention A (groups 2 and 3) to all those who did not (groups 1 and 4). Similarly, the effect of intervention B is determined by comparing all patients who received intervention B (groups 2 and 4) to all patients who did not (groups 1 and 3). This so-called ‘at the margins’ analysis assumes the independent effect of the interventions and allows for the factorial design to provide a similarly powered study with fewer patients than two separate clinical trials of the two interventions. This simple version of a factorial design is also sometimes referred to as a ‘dismantling design’ due to its ability to deconstruct elements of a multifactor intervention.
The treatment effects of both interventions should be additive when doing an ‘at the margins’ analysis, meaning no interaction was present. When the treatment effect is less than (antagonistic) or more than additive (synergistic), then an interaction was present between the two treatments and the ‘at the margins’ analysis is inappropriate. The interaction causes an inaccurate estimate of the individual effect of each intervention. In the presence of an interaction, the analysis must be performed ‘inside the table’, meaning the effect of intervention A is determined by comparing patients who received only intervention A (group 3) to those who received neither intervention (group 1). Similarly, the effect of intervention B is determined by comparing patients who received only intervention B (group 4) compared with those who received neither intervention (group 1). Patients in group 2 are excluded. This results in fewer observations in the statistical analyses, lower power, and more uncertainty in the estimate of treatment effect.
An example of a factorial study was the Fluid Lavage of Open Wounds (FLOW) trial.23 The FLOW study was a 2×3 factorial trial that examined what type of fluid (intervention) and what degree of pressure (factor with multiple levels) were used to irrigate open fractures. This study had two interventions—soap or normal saline as the irrigant used and three degrees of pressure—low, gravity flow, or high pressure.24
Factorial designs allow for the efficient study of multiple interventions assuming they act independently and no interactions are present (table 7). It is rare, particularly in trauma, to deliver one intervention separate from others that will not collectively have an impact on the patient and will not interact with each other. It can also often be impossible to accurately ascertain whether an interaction will be present. Despite the decrease in power associated with identifying an interaction after a factorial trial, the results can still provide important information such as assessing the magnitude of the interaction and information to allow for planning and powering a future clinical trial to determine the independent effect of the intervention of interest.25
Factorial trial design
Bayesian methods
Bayesian methods are not a form of alternative clinical trial design, but rather a different method of inference. While conventional frequentist statistical methods have been used almost universally in the analysis of scientific studies, they have major limitations when used to provide inference in clinical trials. First, they do not provide a clinically applicable answer to the question clinicians find most relevant: What is the probability that the alternate hypothesis is true? Second, they lead to erroneous binary thinking regarding an intervention. Third, they are often misused, misunderstood, and, thus, misinterpreted.26 Finally, they ignore the prior evidence about the intervention, leaving the reader on their own to integrate the results of the study with the results of prior studies.
As a community, we have evolved to reporting the results of a clinical trial as positive or negative (p<0.05 vs. p>0.05), rather than interpreting the more precise measure of inference in its continuous form (eg, p=0.07). This has become common because p values are difficult to interpret and even more difficult to apply clinically. For example, in a frequentist analysis, the alternative hypothesis might be: Patients in hemorrhagic shock who receive drug A will have at least 5% reduction in mortality at 30 days compared with those who do not receive drug A. The null hypothesis will be that there is no difference in 30-day survival. In a frequentist analysis the null hypothesis is assumed to be true and the p value can only provide information relative to this assumption. The p value is the probability of finding the observed data, or more extreme, assuming that the null hypothesis is true. The p value that is generated cannot estimate the probability of the alternative hypothesis being true because the null and the alternative cannot be true at the same time.27 Bayesian methods bridge this gap by providing the more clinically meaningful outcome, the posterior probability, which clinicians can use to perform risk-benefit assessment of an intervention. Simply put, Bayesian methods speak the clinician’s language.
Bayesian methods are similar to how surgeons approach everyday clinical scenarios. When making decisions, we use our experience, prior knowledge, and training to assess the probability that one of many treatments will yield the outcome of interest. We then perform that treatment and assess the outcomes. If the treatment is successful, we increase our probability that the treatment is appropriate in that clinical scenario. If the treatment is unsuccessful, we decrease our probability that the treatment was the best option.
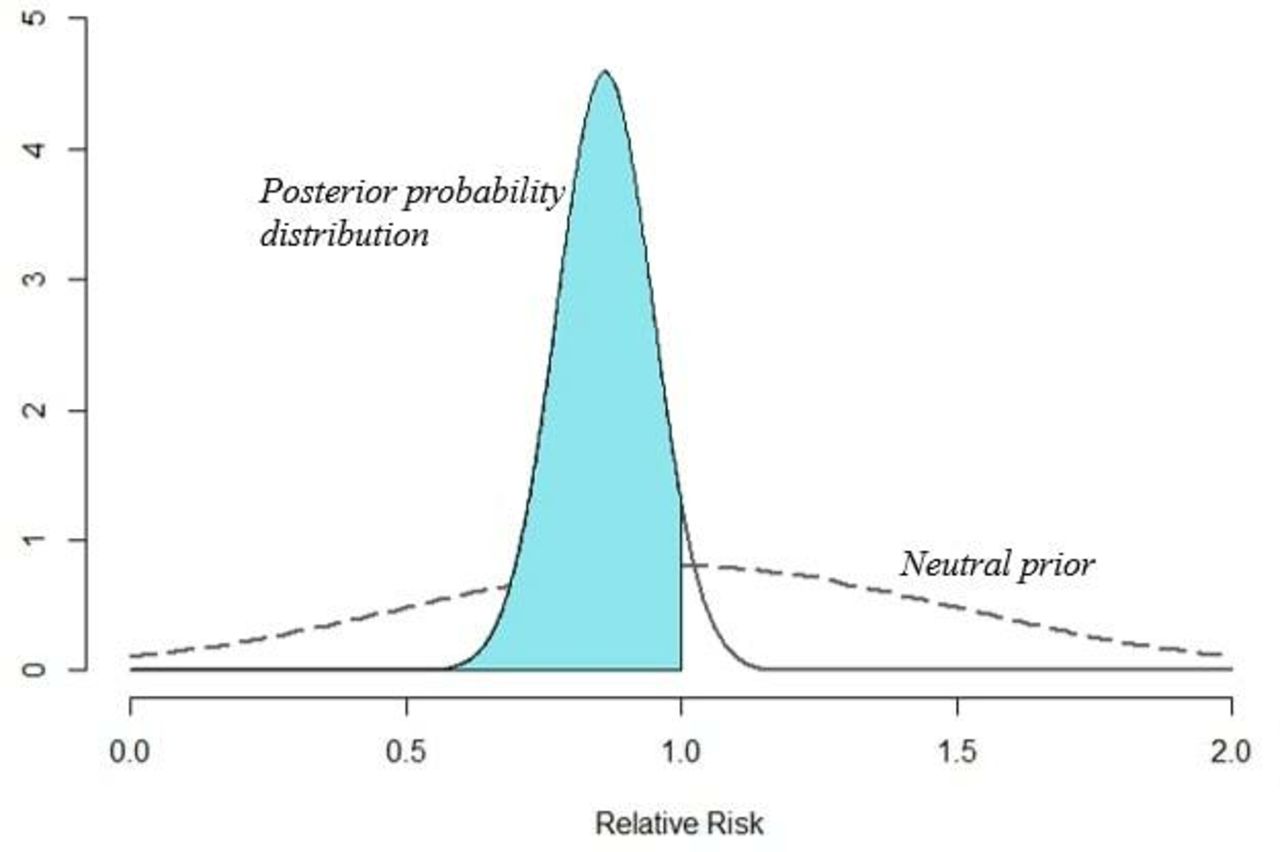
Bayesian methods weigh the degree of uncertainty regarding the effect size of the treatment and combine that with the probability the treatment will be beneficial or harmful. Bayesian analyses explicitly incorporate prior knowledge into the estimates of the probability of treatment effect before the study (the prior probability). When mathematically formalized, Bayes’ theorem provides an optimized rule for updating these prior probabilities with newly observed data. The result is a posterior probability distribution of treatment effect, quantifying the plausibility of both the null and alternate hypotheses (figure 2). This posterior probability statement might take the form: ‘The chance that treatment confers benefit of some magnitude or higher is X%.’ This posterior probability is more intuitive than the analogous interpretation of a p value: ‘Assuming that the null hypothesis regarding treatment is true, the chance of observing the current data, or data more extreme is Y%.’ Note that the former approach provides a direct assessment of the alternative hypothesis (the reason the clinician is engaging in a given course of action). The latter approach makes no direct mention of the alternative hypothesis as frequentist methods result in a non-comparative value that is only in relation to the null hypothesis.

{kind=link}
{kind=link}
Graphical representation of a hypothetical study using Bayesian methods. Consider a trial of two treatments in which the rate of mortality was 34% (112/331) in treatment A and 40% (134/333) in treatment B. Frequentist inference would provide a risk ratio of 0.84 (95% CI 0.69 to 1.03, p=0.09). The result would be stated that no statistically significant difference between these two interventions was observed. In contrast, a Bayesian analysis, using a vague, neutral prior, would provide a risk ratio of 0.86% and 95% credible interval of 0.70–1.04. Plotting this posterior distribution would result in an area under the curve to the left of 1 (ie, decreased mortality) of 94% of the entire distribution. The result would be stated as such: there was a 94% probability that treatment A reduced mortality compared with treatment B.
In addition to providing clinically applicable answers to the question posed, Bayesian methods have other advantages in clinical trials. First, the degree of uncertainty of the treatment effect is built into the posterior probability. Second, prior knowledge is explicitly included into the estimates of the probability of treatment effect. This inclusion allows for iterative updating of the posterior probability. Third, interim analyses under the frequentist method inflate the overall type I error rate requiring sequential methods to address (‘alpha spending’). Bayesian methods have no such penalty and allow for a monitoring schedule at any stage and with any cohort size.
The main disadvantage to using Bayesian methods is the determination of the prior probability. When no data exist, the choice of a prior can be subjective and greatly affect the posterior probability. Ideally, the prior is based on already published high-quality studies when available. In scenarios where no prior data exist, conservative analyses using neutral priors with a range limited to plausible effect sizes may be used, another advantage of Bayesian methods. Alternatively, Bayesian methods permit a sensitivity analysis whereby a range of plausible prior probabilities are provided and the clinician can consider the prior they think credible.
Conclusions
Despite many advances in the care of injured patients during the last several decades, many unknowns remain. Traditional RCTs may be impractical in the injured patient population. Alternative trial designs may offer a way to move the field of injury research forward more rapidly if they can be widely applied to the many research questions that remain to be answered. Further, relying solely on frequentist methods of statistical inference may result in dismissing the results from small clinical trials or from trials that are deemed negative based on an arbitrary binary interpretation of a p value.
Acknowledgments
The authors greatly appreciate the ongoing financial support of the Coalition for National Trauma Research Scientific Advisory Committee (CNTR-SAC) from the following organizations: American Association for the Surgery of Trauma (AAST), American College of Surgeons Committee on Trauma (ACS-COT), Eastern Association of the Surgery of Trauma (EAST), National Trauma Institute (NTI), and Western Trauma Association (WTA).
References
Footnotes
Collaborators Members of the Coalition for National Trauma Research Scientific Advisory Committee: Saman Arbabi, MD FACS, Eileen M. Bulger, MD FACS (Department of Surgery, University of Washington); Mitchell J. Cohen, MD FACS (Department of Surgery, University of Colorado); Todd W. Costantini, MD FACS (Department of Surgery, UC San Diego School of Medicine); Marie M. Crandall, MD, MPH FACS (Department of Surgery, University of Florida College of Medicine Jacksonville); Rochelle A. Dicker, MD FACS (Departments of Surgery and Anesthesia, UCLA Geffen School of Medicine); Elliott R. Haut, MD, PhD FACS (Departments of Surgery, Anesthesiology and Critical Care Medicine and Emergency Medicine, The Johns Hopkins University School of Medicine; The Armstrong Institute for Patient Safety and Quality, Johns Hopkins Medicine; Department of Health Policy and Management, The Johns Hopkins Bloomberg School of Public Health); Bellal Joseph, MD FACS (Department of Surgery, University of Arizona); Rosemary A. Kozar, MD, PhD FACS (Department of Surgery, University of Maryland); Ajai K. Malhotra, MD FACS (Department of Surgery, University of Vermont); Avery B. Nathens, MD, PhD, FRCS, FACS (Department of Surgery, University of Toronto); Raminder Nirula, MD, MPH FACS (Department of Surgery, University of Utah); Michelle A. Price, PhD, MEd (National Trauma Institute, UT Health - San Antonio); Jason W. Smith, MD FACS (Department of Surgery, University of Louisville); Deborah M. Stein, MD, MPH FACS FCCM (Department of Surgery, University of California - San Francisco); Ben L. Zarzaur, MD, MPH FACS (Department of Surgery, University of Wisconsin School of Medicine and Public Health).
Contributors All authors (JAH, BLZ, RN, BTK, AKM) made substantial contributions to the design, drafting, and critical revisions of the work. All authors also gave final approval of the version submitted.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; internally peer reviewed.