Article Text

Abstract
Background Damage control laparotomy (DCL) is an abbreviated operation intended to prevent the development of hypothermia, acidosis, and coagulopathy in seriously injured patients. The indications for DCL have since been broadened with no high-quality data to guide treatment. For patients with an indication for DCL, we aim to determine the effect of definitive laparotomy on patient morbidity.
Method This is a pragmatic, parallel-group, randomized controlled pilot trial. Emergent laparotomy is defined as admission directly to the operating room from the emergency department within 90 min of arrival. DCL indications excluded from the study include packing of the liver or retroperitoneum, abdominal compartment syndrome prophylaxis, to expedite interventional radiology for hemorrhage control, and the need for ongoing transfusions and/or continuous vasopressor support. When a surgeon determines a DCL is indicated, the patient will be screened for inclusion and exclusion criteria. Patients with any indication for DCL that is not excluded are eligible for randomization. Patients will be randomized intraoperatively to DCL (control) or definitive fascial closure of the laparotomy (intervention). The primary outcome will be major abdominal complication or death within 30 days. Major abdominal complication is a composite outcome including fascial dehiscence, organ/space surgical site infection, enteric suture line failure, and unplanned reopening of the abdomen. Outcomes will be compared using both frequentist and Bayesian statistics.
Discussion In patients with an indication for DCL, this trial will determine the effect of definitive laparotomy on major abdominal complications and death and will inform clinicians on the risks and benefits of this procedure. Regardless of the study outcome, the results will improve the quality of care provided to injured patients.
Trial registration number NCT02706041.
- laparotomy
- damage control
- randomized clinical trial
- complications of laparotomy
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Abbreviation of a trauma laparotomy to control hepatic bleeding or after the onset of coagulopathy was first described in 1908 and became acceptable in the 1970s and 1980s.1–4 The term ‘damage control’ was used to describe a type of abbreviated laparotomy for trauma in the 1990s.5 Dissemination and implementation of damage control laparotomy (DCL) occurred rapidly with centers across the USA publishing reports of their experience with this intervention.6 ,7
As comfort with the open abdomen necessitated by DCL increased and as temporary abdominal closure devices improved,8 ,9 the indications for DCL became more liberal.10 ,11 The morbidity associated with DCL also became more evident, including incisional hernia formation, enterocutaneous and enteroatmospheric fistula formation, superficial and organ/space surgical site infections, organ failure, and fascial dehiscence.12–16
The absolute and relative clinical indications that have evolved over time are neither well defined nor supported by any high-quality data. Indeed, expert opinion continues to be the major determinant of appropriateness of select indications.17 ,18 The majority of trauma surgeons acknowledge that DCL is a necessary tool for select patients despite the lack of data. A major obstacle to performing clinical trials of DCL is surgeon equipoise. This protocol uses the information gathered from a 2-year quality improvement project at our institution in which a number of indications for DCL were identified to be ones for which surgeons have clinical equipoise.
In this parallel group, randomized controlled trial, we compare the treatment effect of DCL versus definitive laparotomy on major abdominal complications (MAC) or death. We hypothesize that definitive laparotomy will result in lower MACs or death compared with DCL. The following describes the design, rationale, and implementation of the DCL trial, the first clinical trial on the effectiveness of DCL following emergent trauma laparotomy.
Methods/design
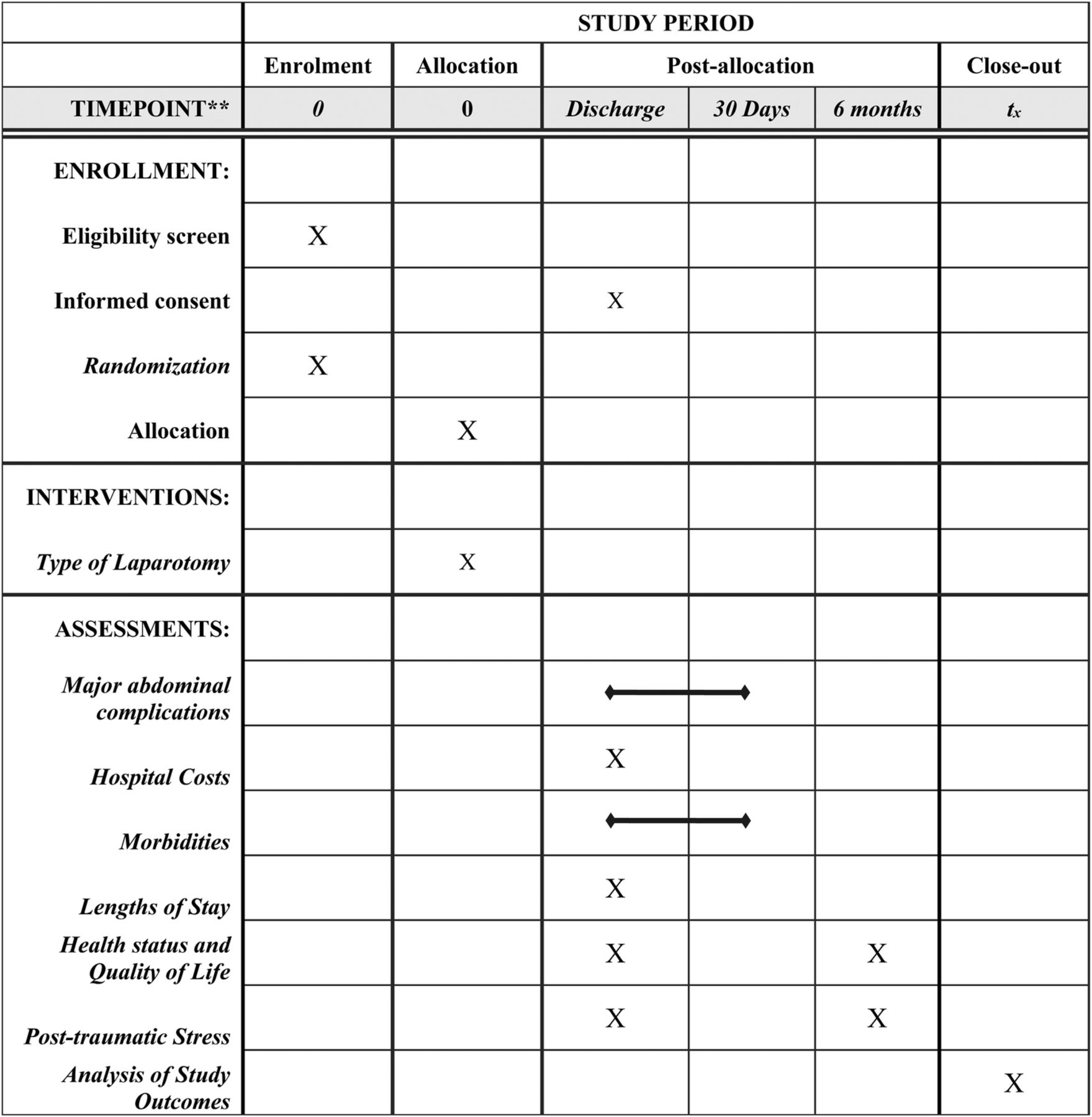
The DCL trial is a single-center, randomized, controlled, pilot trial of trauma patients undergoing emergent laparotomy following injury. This manuscript was written in accordance to the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) 2013 guidelines.19 A SPIRIT diagram detailing the timing of screening, randomization, allocation, and assessment of outcomes is provided in figure 1.
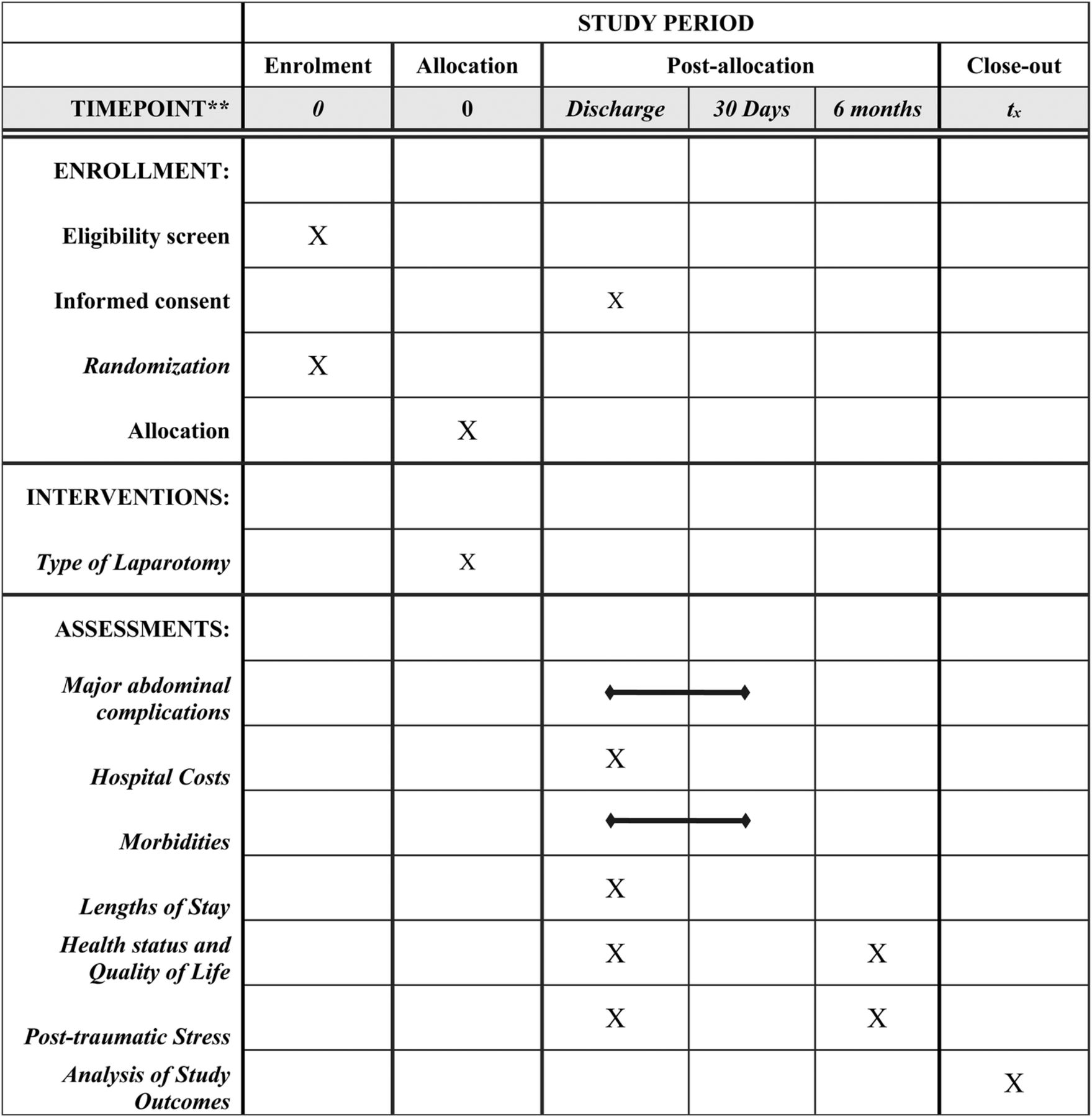
SPIRIT diagram. The figure details the timing of enrollment activities, intervention allocation, and assessments of outcomes over the course of the clinical trial. SPIRIT, Standard Protocol Items: Recommendations for Interventional Trials.
Study setting
This study is being conducted at the Red Duke Trauma Institute at Memorial Hermann Hospital-Texas Medical Center. The Red Duke Trauma Institute is one of two level 1 trauma centers in the Houston, Texas metropolitan area. The trauma center's urban location combined with the fact that it is the city's only trauma center that receives trauma patients via aeromedical transport results in a broad mix of urban and rural patients suffering penetrating and blunt trauma.
Eligibility criteria
All adult trauma patients (≥16 years of age) undergoing emergent trauma laparotomy are screened for possible inclusion into the DCL trial (table 1). Emergent laparotomy is defined as admission directly to the operating room from the emergency department in ≤90 min from initial hospital arrival. The trigger to potentially enroll the patient is the attending surgeon's determination during laparotomy that the patient has an indication for DCL.
Inclusion and exclusion criteria for the DCL trial
The major hurdle to any clinical trial of DCL is surgeon equipoise. Prior to starting the DCL trial, a 2-year, prospective quality improvement project was performed at our institution. One of the aims of the project was to determine indications for DCL for which surgeons had equipoise. Indications for DCL which lacked surgeon equipoise to perform definitive laparotomy included: packing of the liver or retroperitoneum for hemorrhage control; expedited transfer to interventional radiology for hemorrhage control; abdominal compartment syndrome treatment or prophylaxis; and continuous vasopressor use, ongoing transfusions, and/or persistent hypotension. These indications are exclusion criteria for the DCL trial.
Indications for DCL which group equipoise for definitive laparotomy was found included: planned second-look laparotomy; expedited transfer of the patient for postoperative imaging (eg, CT of the head to diagnose a traumatic brain injury) or intensive care; isolated acidosis without ongoing transfusions or continuous vasopressor use; and contamination. These indications are inclusion criteria for the DCL trial. Any other indication not specifically excluded above will be eligible for randomization, such as prehospital/emergency department hypotension, hypothermia, injury patterns, operative time, estimated blood loss, and volume of resuscitation.18 As this is a pilot study, the identification of additional indications for DCL that lack surgeon equipoise will help to plan a larger, multicenter study.
Interventions
Subjects are randomized during their emergent trauma laparotomy into one of two groups: definitive laparotomy (intervention—completion of all portions of the laparotomy and fascial closure) or DCL (control—completion of necessary portions of the laparotomy and temporary abdominal closure) (figure 2). The choice of temporary abdominal closure will not be controlled; however, the usual institutional practice is to use the KCI ABThera Open Abdomen Negative Pressure Therapy System.

{kind=link}
{kind=link}
Flow chart for the DCL trial. Patients with an indication for DCL in which there is surgeon equipoise will be randomized to DEF (intervention) or DCL (control). DCL, damage control laparotomy; DEF, definitive laparotomy.
To increase surgeon enrollment and randomization of patients, daily screening of all emergent trauma laparotomies is being performed to provide real time audit and feedback to the surgeons at the trauma center. Additionally, the inclusion and exclusion criteria were selected based on surgeon feedback during the preceding quality improvement project to ensure equipoise for randomization. Having included the participating surgeons as stakeholders during the creation of the study protocol should help to improve surgeon enrollment and adherence.
Other than randomization allocation, all other clinical treatments are performed according to institutional protocols and usual practice. Patients with multisystem injuries are included and clinical care and management of extra-abdominal injuries (eg, external fixation of long bone fractures, usage of temporary intra-arterial vascular shunts) are left to the discretion of the operating surgeon.
Outcomes
The primary outcome of the trial is MAC or death within 30 days of laparotomy. MAC is a binary, composite outcome consisting of any of the following: organ/space surgical site infection, enteric suture line failure, fascial dehiscence, or unplanned return to the operating room for an abdominal complication. Organ/space surgical site infection is defined in accordance to Centers for Disease Control and Prevention guidelines.20 Enteric suture line failure (enteric anastomotic leak) is defined as leakage of enteric contents from a gastrointestinal anastomosis with or without the need for reoperation. Fascial dehiscence is defined as separation of closed fascia with or without evisceration. Unplanned reoperation for abdominal complication is defined as reopening of previously closed fascia for any intra-abdominal complication.
Secondary outcomes include non-abdominal morbidities, hospital, intensive care unit, and ventilator-free days (equal to 30 minus the total hospital/intensive care unit/ventilator days with >30 or death equal to a 0 value), total hospital stay costs, and patient-centered outcomes.
Non-abdominal complications will be identified based on standardized definitions used in the National Trauma Databank and include: acute kidney failure, adult respiratory distress syndrome, deep venous thrombosis, pulmonary embolism, pneumonia, and urinary tract infection. Per-patient cost information will be obtained from the hospital and used to study the healthcare resource usage of DCL.
For patient-centered outcomes, each patient's health status will be queried at discharge and 6 months after discharge using the Standard Gamble and EuroQol-5D(5L).21 ,22 The Posttraumatic Stress Disorder Check List-Civilian will be administered 6 months after discharge by phone interview. Additionally, time to return to work will be obtained at the 6-month interview.
Sample size
The study aims to enroll 56 patients, 28 in each group. This is based on: (1) unpublished, preliminary data from the quality improvement project showing a MAC or death rate of 55% in patients undergoing DCL who may have safely undergone definitive laparotomy and 18% in patients undergoing definitive laparotomy; (2) an α of 0.05, (3) 80% power; and (4) a 10% drop out rate.
Randomization
Allocation is occurring through sequentially numbered, opaque, sealed envelopes kept in the research assistants' office and opened in the operating room. An independent statistician determined the randomization sequence and an uninvolved administrative assistant labeled the cards and envelopes.
A 1:1 allocation ratio using a permuted block design of 4 or 6 was used to ensure equal number of patients in each group.
Randomization occurs during the emergent trauma laparotomy. Research assistants are in the hospital 24 hours a day, 7 days a week. The research assistant will ask the attending trauma surgeon periodically throughout emergent laparotomies to determine if the patient meets eligibility criteria. All trauma surgeons at the center have agreed to participate. If the patient meets all inclusion criteria and has no exclusion criteria, the research staff will open the opaque envelope and notify the attending surgeon to which group the subject has been randomized.
Blinding
Blinding of the clinical staff responsible for providing care to the enrolled patients is not possible. To address this limitation, the individual components of the composite, primary outcome MAC or death have been objectively defined. In situations where the determination of a MAC is unclear, an uninvolved surgeon from the elective general surgery division who does not provide care for trauma patients will adjudicate the presence or absence of a MAC. Secondary outcomes will be defined according to National Trauma Databank standards and retrieved from the institutional trauma registry.
Data collection, management, integrity and confidentiality
Data are collected via direct observation by study staff on standardized case report forms. Direct observation continues until either the patient is determined to not be eligible for the trial or the completion of the laparotomy. Data are then entered into REDCap a web-based data management application. Each item on the web form has validity checks performed to ensure that the data entered are accurate and that items are not skipped during entry by mistake. Checks have been developed by the principal investigator, the study statistician, and the research nurse. Entered data are audited weekly by the principal investigator and research nurse to ensure accuracy.
All hard copy source documentations are kept in a secured, locked cabinet in the research coordinator's office. All study documents will be maintained in a secure location for 2 years following study completion.
Analysis
The number of screened patients and reasons for exclusion will be reported. Protocol violations and reasons for those violations will be reported and detailed. Differences in primary and secondary outcomes across treatment groups will be compared on an intent-to-treat basis using the Wilcoxon rank-sum test, Pearson's χ2 test, and Fisher's exact test, for continuous, binary, and sparse binary outcomes, respectively.
Given the small sample size of the trial, we will augment the frequentist model described above with a Bayesian analysis.23 We will use a log binomial model to compute the relative risk of the primary outcome with DCL compared with definitive laparotomy. The Bayesian analysis will use a negative prior centered at a relative risk of 1.0. The Bayesian analysis will provide the probability that definitive laparotomy decreases the incidence of MAC or death compared with DCL. Bayesian statistics is a helpful method to describe and understand clinical trials in which there is uncertainty in the baseline treatment effect of an intervention and those with a small sample size.
Data and safety monitoring board
To assess for harm, blinded, univariate outcomes between the two groups will be assessed every 6 months throughout the study period by a data and safety monitoring board (DSMB), composed of a surgical oncologist, a pediatric surgeon, an anesthesiologist, and an independent statistician.
At the first meeting following 50% recruitment, a formal Bayesian interim analysis will be performed and presented to the DSMB to assess the probability of a beneficial or harmful effect of definitive laparotomy on MAC or death. The DSMB will be instructed to recommend stopping the trial if the interim Bayesian analysis suggests a >85% probability of harm for the intervention.
Research approval
The Institutional Review Board (IRB) of the University of Texas Health Sciences Center at Houston (UT Health) approved the study protocol on June 3, 2016 following satisfactory community consultation and public notification. Enrollment began on July 7, 2016 and is scheduled to continue for 2 years.
Informed consent
Given the emergent nature of the intervention—a trauma laparotomy—individual informed consent is not possible prior to enrolling patients. Experience with legally authorized representatives has shown that they are not available for many hours after admission.24 Exception from informed consent (EFIC) allows subjects to be randomized before they or their legally authorized representative are consented. The use of EFIC requires both public notification and community consultation prior to starting the trial. The IRB at UT Health approved this trial using EFIC with delayed consent of the patient or the patient's legally authorized representative.
Discussion
The DCL trial is a pilot, single-center clinical trial which aims to determine the effect of DCL compared with definitive laparotomy on MAC and/or death. This pilot study will provide the most valid estimate of treatment effect to date on the effect of definitive laparotomy and DCL. This protocol has unique features to help address the many potential difficulties in performing a clinical trial in critically injured patients undergoing emergency surgery.
First, while the Division of Acute Care Surgery at the University of Texas McGovern Medical School has an excellent record of trauma surgeon buy in for well-designed randomized controlled trials,25 ,26 it remains a concern that surgeons will choose not to enroll potentially eligible patients or fail to implement the randomized treatment. To mitigate this concern, several processes have been implemented. One includes the preliminary work during a 2-year quality improvement project which used surgeon input as stakeholders to determine indications for which they had equipoise to randomize. Additionally, damage control laparotomies will be discussed the following day to assess eligibility. If a patient is felt to have met the inclusion criteria, the principal investigator will provide near real-time surgeon feedback to improve future compliance.
Second, the indications that lack surgeon equipoise at this institution may be different than indications at other institutions. Thus, the external validity of the study is in question. Nevertheless, this is a pilot study and will be able to provide the most valid estimate of treatment effect of definitive laparotomy and DCL to date as all other studies on the subject are retrospective.
Third, though the sample size is small (56 patients), given the emergent nature of the enrollment and potential difficulty in surgeons agreeing to randomization, it is possible that the number of patients enrolled at the end of the trial period will be less than anticipated. Our institution has high volume, with ∼220 emergency trauma laparotomies per year with preliminary data suggesting 24% (53 patients per year) of all patients will be eligible for enrollment. In order to address this potential impediment, funding has been secured for the trial to run for 2 years and we will use Bayesian statistics to augment our frequentist statistics.
In summary, the DCL trial will be the first randomized clinical trial of definitive abdominal closure and DCL in trauma patients. As current reports of morbidity following DCL are retrospective, this will provide the least biased estimates of treatment effects for definitive laparotomy and DCL.
References
Footnotes
Contributors JAH, JP, LEV, EEF, LJM, BAC, and JBH made substantial contributions to the concept and design of the clinical trial, were involved in the drafting and revising of the manuscript, gave final approval of the manuscript to be published, and agreed to be accountable for all aspects of the work.
Funding JAH is supported by the Center for Clinical and Translational Sciences, which is funded by the National Institutes of Health Clinical and Translational Award KL2 TR000370 from the National Center for Advancing Translational Sciences.
Disclaimer The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Advancing Translational Sciences or the National Institutes of Health.
Competing interests None declared.
Ethics approval The Institutional Review Board (IRB) of the University of Texas Health Sciences Center at Houston (UT Health) approved the study protocol.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.