Article Text

Abstract
Background Resuscitative Endovascular Balloon Occlusion of the Aorta (REBOA) increases cardiac-afterload and is used for patients in hemorrhagic shock. The cardiac tolerance of prolonged afterload augmentation in this context is unknown. The aim of this study is to quantify cardiac injury, if any, following 2, 3 and 4 hours of REBOA.
Methods Anesthetized swine (70–90 kg) underwent a 40% controlled hemorrhage, followed by supraceliac resuscitative endovascular balloon occlusion of the aorta (REBOA) for 2 (n=5), 3 (n=5), and 4 hours (n=5). High-fidelity arterial wave form data were collected, and signal processing techniques were used to extract key inflection points. The adjusted augmentation index (AIx@75; augmentation pressure/pulse pressure, normalized for heart rate) was derived for use as a measure of aortic compliance (higher ratio = less compliance). Endpoints consisted of electrocardiographic, biochemical, and histologic markers of myocardial injury/ischemia. Regression modeling was used to assess the trend against time.
Results All animals tolerated instrumentation, hemorrhage, and REBOA. The mean (±SD) systolic blood pressure (mm Hg) increased from 65±11 to 212±39 (p<0.001) during REBOA. The AIx@75 was significantly higher during REBOA than baseline, hemorrhage, and resuscitation phases (p<0.05). A time-dependent rise in troponin (R2=0.95; p<0.001) and T-wave deflection (R2=0.64; p<0.001) was observed. The maximum mean troponin (ng/mL) occurred at 4 hours (14.6±15.4) and maximum T-wave deflection (mm) at 65 minutes (3.0±1.8). All animals demonstrated histologic evidence of acute injury with increasing degrees of cellular myocardial injury.
Discussion Prolonged REBOA may result in type 2 myocardial ischemia, which is time-dependent. This has important implications for patients where prolonged REBOA may be considered beneficial, and strategies to mitigate this effect require further investigation.
Level of evidence II.
- aortic occlusion
- resuscitative endovascular balloon occlusion of the aorta
- reboa
- hemorrhage
- cardiac
- cardiac injury
- ischemia
- arterial waveform
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- aortic occlusion
- resuscitative endovascular balloon occlusion of the aorta
- reboa
- hemorrhage
- cardiac
- cardiac injury
- ischemia
- arterial waveform
Introduction
Hemorrhage is the leading cause of preventable deaths after traumatic injury.1 Resuscitative endovascular balloon occlusion of the aorta (REBOA) is a hemorrhage control adjunct which allows for the attenuation of hemorrhage by decreasing distal blood flow and increasing afterload and blood flow proximally.2 3 In an effort to improve outcomes, as well as broaden the clinical applications of REBOA, there has been substantial interest in performing REBOA closer to the point of injury,4 5 and for prolonged durations,6 to mitigate extended transport time or facilitate interhospital transfer.
This has already led to small case series and reports of clinicians performing prehospital REBOA in both military4 and civilian settings,5 as well as with interhospital transfers.7 Previous studies have focused on the effects of the inflammatory and distal ischemic burden that develops with prolonged aortic occlusion (AO)6 8 9; however, the physiologic effects of REBOA for extended periods of time on the myocardium remain ill-defined.
Whereas animal models10 11 of cardiac arrest have shown the benefit of AO on coronary perfusion and increased rates of return of spontaneous circulation, the tolerance and effect of AO on the non-arrested heart are less well described. A severe abrupt increase in afterload alone has been described to cause type 2 myocardial ischemia/injury, such as with cases of hypertensive emergency related to pheochromocytomas, medication side effects, and during aortic cross-clamping.12–14 Others15 have demonstrated that there is a significantly attenuated afterload response to AO with decreased preload (hemorrhage) in a swine model which may result in a cardioprotective effect; however, this has not been well characterized.
The objective of this study was to characterize the effects of extended durations of REBOA on the heart in the context of severe hemorrhage in a non-diseased animal model.
Materials and methods
Study overview
This in vivo study characterizes the effect of hemorrhagic shock and AO by REBOA on the heart. Yorkshire swine (Sus scrofa) weighing between 70 and 90 kg were used. This study consisted of four phases: preparation, hemorrhage, AO, and critical care. This study design was chosen to simulate prolonged prehospital REBOA close to the point of injury. The hemorrhage phase represents the point of injury before REBOA is deployed, and the final phase consists of definitive care with resuscitation.
Animal preparation and hemorrhage
General anesthesia was induced using intramuscular ketamine (10–15 mg/kg) and xylazine (1–2.2 mg/kg), followed by intravenous propofol and maintained with isoflurane (range 1%–4%) by mask and subsequently by tracheostomy. Animals were ventilated using a volume-controlled mode of 6 cc/kg with a fractional inspired oxygen of 40% to 100% to maintain peripheral capillary oxygen saturation >92%.
Surgical exposure via a midline neck incision was performed to cannulate the common carotid and internal jugular veins. An arterial line was placed in the left common carotid artery, and a flow probe (FSB-Series, Transonic Systems, Ithaca, NY) was placed distal to the arterial line. The jugular veins were also cannulated bilaterally to permit intravenous access and placement of a Swan-Ganz catheter. The right femoral vein was cannulated via a cut-down to permit venesection. The right femoral artery was cannulated with a 7 Fr sheath, and an ER-REBOA catheter (Prytime Medical, Boerne, TX) was advanced into zone 1 of the aorta. An open cystostomy was performed for urine drainage.
Volume-controlled hemorrhage
Hemorrhagic shock was induced using a volume-controlled exponential venesection technique. Assuming a porcine blood volume of 66 mL/kg, 40% of the animal’s blood volume was removed over the course of 20 minutes from the femoral venous catheter. The first 20% of blood volume was removed over 7 minutes, and the remaining 20% of blood volume was removed over 13 minutes. Animals then underwent a 10-minute period where no intervention or resuscitation was performed.
Aortic occlusion
Zone 1 AO was performed at the end of the hemorrhage protocol, which was designated T0. Complete AO was confirmed using fluoroscopy. The study was then divided into three groups undergoing 2 hours of occlusion (2-HR-REBOA; n=5), 3 hours (3-HR-REBOA; n=5), and 4 hours (4-HR-REBOA; n=5).
Resuscitation/Critical care
A period of resuscitation ensued for 1 hour after balloon deflation before euthanasia and necropsy. This commenced with the return of the venesected blood, which had been collected in citrated blood and was coadministered with intravenous calcium. Once this was exhausted, intravenous 0.9% saline was administered when mean arterial pressure was <60 mm Hg, until the animal was no longer fluid-responsive, whereupon an infusion of norepinephrine was commenced. Hyperkalemia was corrected using boluses of 10 units of insulin and 50% dextrose. Hypoglycemia was treated with intravenous boluses of 50% dextrose.
Arterial wave form assessment of myocardial work and strain
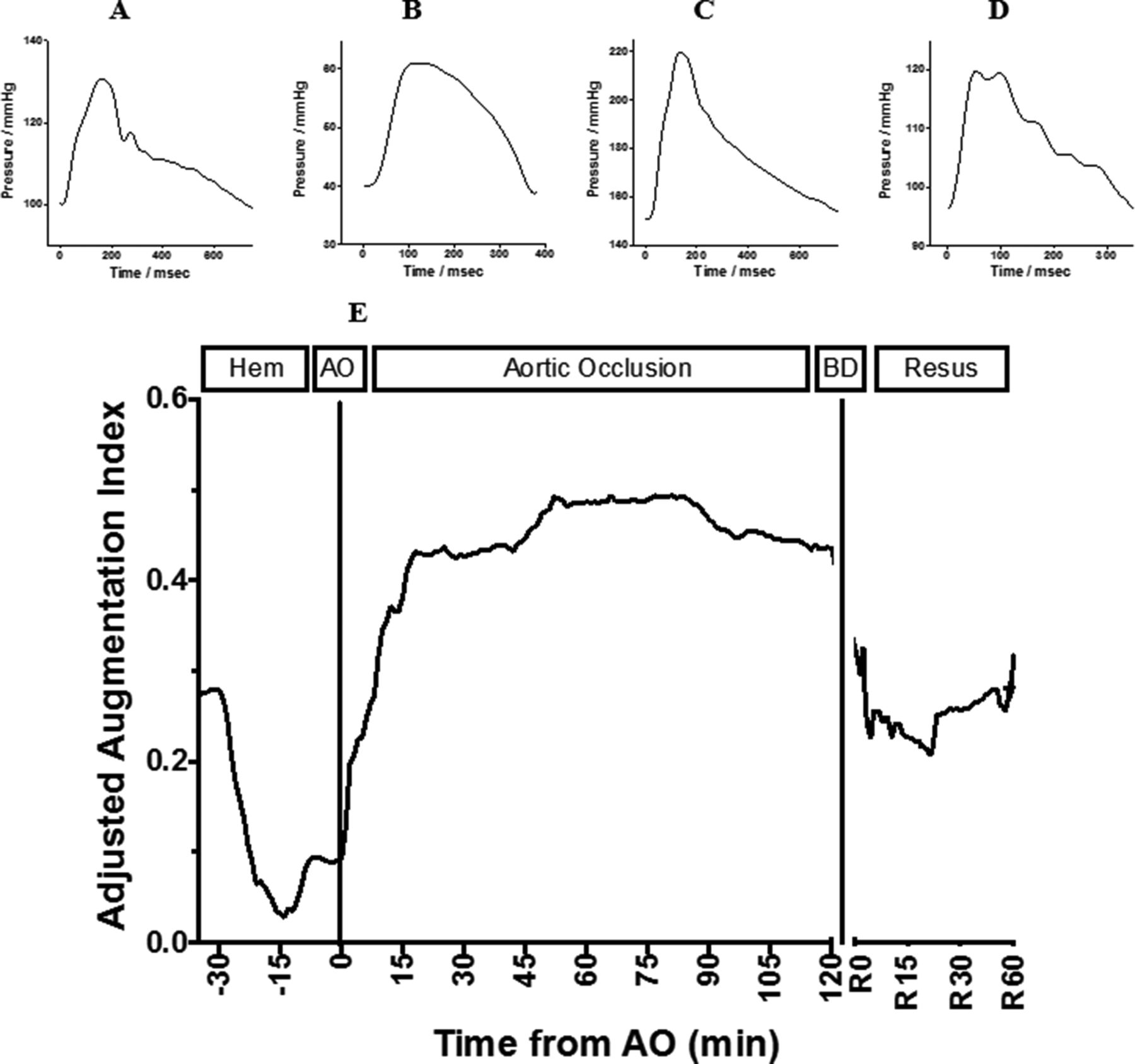
Arterial wave form data were collected continuously from the carotid arterial line at 240 Hz. Analysis was performed using MATLAB (MathWorks, Natick, MA). Specific features (see figure 1) were abstracted from the wave form using an algorithm that identified these features based on the zenith, nadir, and inflection points of the first and second derivatives of the wave form. Representative wave forms at differing time points during the experiment (baseline, immediately after hemorrhage, during AO, and during the resuscitation phase) were obtained to demonstrate the differences in wave form morphology.
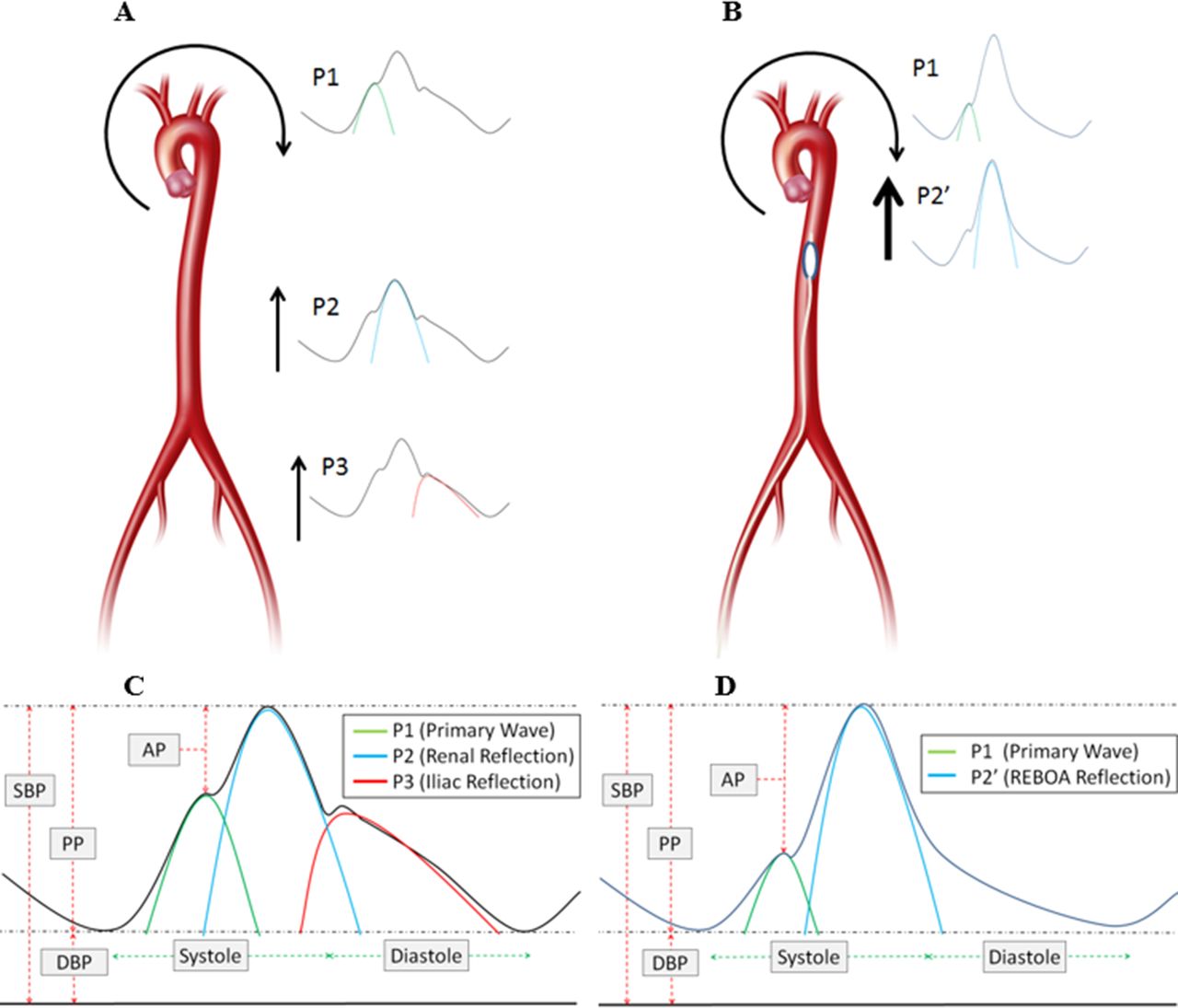
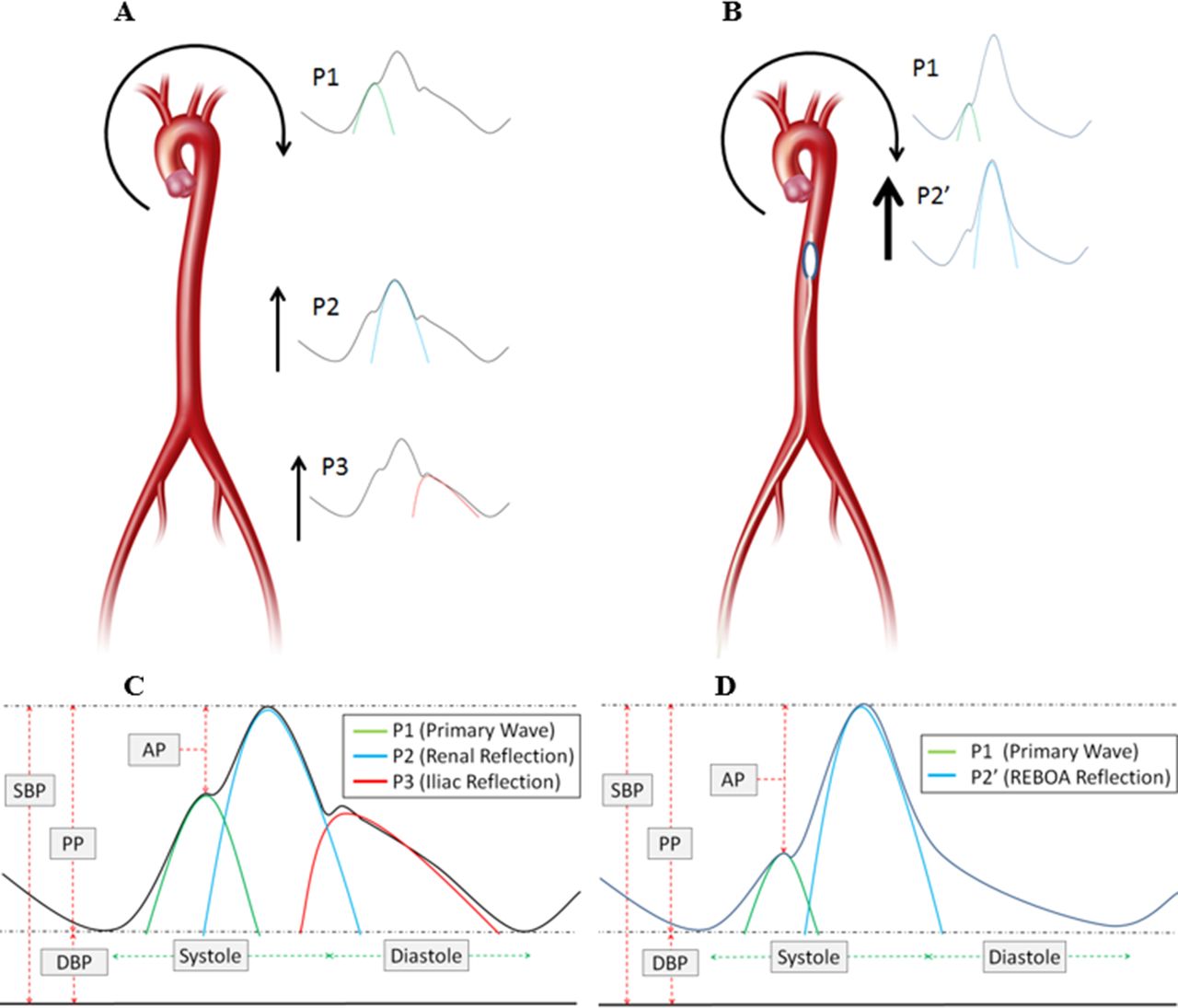
Example of wave components of the arterial wave form and augmentation pressure. A and C demonstrate typical antegrade and reflective wave components. B and D demonstrate antegrade and reflective wave components with REBOA. AP, augmentation pressure; DBP, diastolic blood pressure; PP, pulse pressure; REBOA, resuscitative endovascular balloon occlusion of the aorta; SBP, systolic blood pressure.
The arterial wave form is a composite of multiple pressure waves that are produced either by cardiac contraction or reflection off the distal arterial tree and can be considered to consist of three major waves (figure 1A,C).16 P1 represents the primary antegrade systolic wave, P2 the first wave reflection at the level of the renal arteries, and P3 represents the iliac wave reflection. AO effectively excludes a significant portion of the arterial tree and introduces a more proximal first wave reflection site (figure 1B,D), resulting in an increased augmentation pressure and increase in afterload.17
AO has been demonstrated to increase the augmentation pressure/index (augmentation index; augmentation pressure/pulse pressure),17 and is very similar to those changes caused by aortic stiffening and decreased compliance.18 Importantly, these changes from progressive aortic stiffening have been implicated with increased myocardial strain, oxygen consumption, work, and a decrease in myocardial perfusion pressure.18 In fact, the amount of aortic stiffening (especially the augmentation index) has been closely linked to the severity of heart failure and heart disease.19–21
Quantification of myocardial work and strain throughout the experiments, especially during REBOA, was performed by measuring the unadjusted and adjusted augmentation indices. The augmentation index has been shown to linearly correlate with heart rate, and as heart rate increases the augmentation index decreases.22 23 To account for this, the adjusted augmentation index normalizes values to a heart rate of 75 (with a correction factor of 4.8% per 10 beats per minute—AIx@75). Of note, this methodology is used in commercial digital pulse analyzers such as SphygmoCor Px (AtCor Medical, Itasca, IL).24
ECG wave form assessment of myocardial ischemia
Three-lead (I, II, III) telemetry was continuously recorded. The telemetry recordings were reviewed by a cardiologist (HB) for signs of ischemia, including ST-segment changes and T-wave changes. T-wave amplitude was measured over the course of the experiments. This metric was specifically chosen as changes in T-wave amplitude have been shown to be an early ECG feature of myocardial ischemia. This is related to decreased action potential durations via ATP-sensitive K+ channels.14 25
Laboratory and histologic assessment of myocardial ischemia
Arterial and venous blood samples were acquired every 30 minutes and tested for changes in serum markers of myocardial injury, specifically troponin. On necropsy, the heart was analyzed for evidence of cellular damage using H&E stain. While non-specific changes such as vascular congestion, nuclear disarray, or vacuolization were noted, the percentage of hypereosinophilia was reported. The severity of hypereosinophilia is an early indicator of coagulative necrosis in myocardial ischemia.26
Endpoints and statistical analysis
The primary endpoint of this study was evidence of myocardial injury as measured by T-wave deflection, troponin, and histologic evidence of cardiac eosinophilia. The secondary endpoints included the hemodynamic changes observed during the experimental phases, including blood pressure, heart rate, pulmonary pressure, augmentation index, and wave form morphology.
Data were organized using Microsoft Excel (Redmond, WA) and analyzed using GraphPad Prism (La Jolla, CA) and MATLAB. Student’s paired t-tests were used to compare normally distributed continuous variables. A one-way analysis of variance (ANOVA) with multiple comparisons using the Bonferroni correction was employed when comparing more than three variables. Linear regression was used to explore trends. Statistical significance was defined as a p value of 0.05 or less, and the analysis was performed using GraphPad Prism.
Results
Baseline characteristics
All animals tolerated instrumentation, hemorrhage, and REBOA. The mean±SD weight (kg) of the swine was 79.5±5.0 and all animals were female. One animal in the 4-HR-REBOA group sustained an ST-elevation myocardial infarction prior to the induction of hemorrhage and was therefore excluded from the study. Laboratory parameters throughout the experiment are detailed in table 1. The animals received a mean±SD of 7.7±2.3 L of crystalloid fluid resuscitation and 4.7±0.5 units of whole blood transfusions.
Laboratory parameters
Hemodynamic effects of hemorrhage and AO
The aggregate hemodynamic trends throughout the experiments can be seen in figure 2. On induction of hemorrhage the animals became hypotensive, and then had a significant response to AO. The mean±SD systolic blood pressure (mm Hg) increased from 65±11 during hemorrhage to 212±39 (p<0.001) immediately after AO (peaking between 7 and 12 minutes after AO). After the immediate increase in blood pressure, the blood pressure rapidly and partially attenuates at approximately 15 minutes, and then continues to slowly attenuate with continued AO (R2=0.933; p<0.001). After balloon deflation, an immediate drop in blood pressure occurred, which recovered within 10 minutes with resuscitation. The pulmonary artery pressure was less labile than the systemic arterial pressure, experiencing a slight decrease with hemorrhage, a slight increase immediately after balloon inflation, and then a larger increase after balloon deflation.
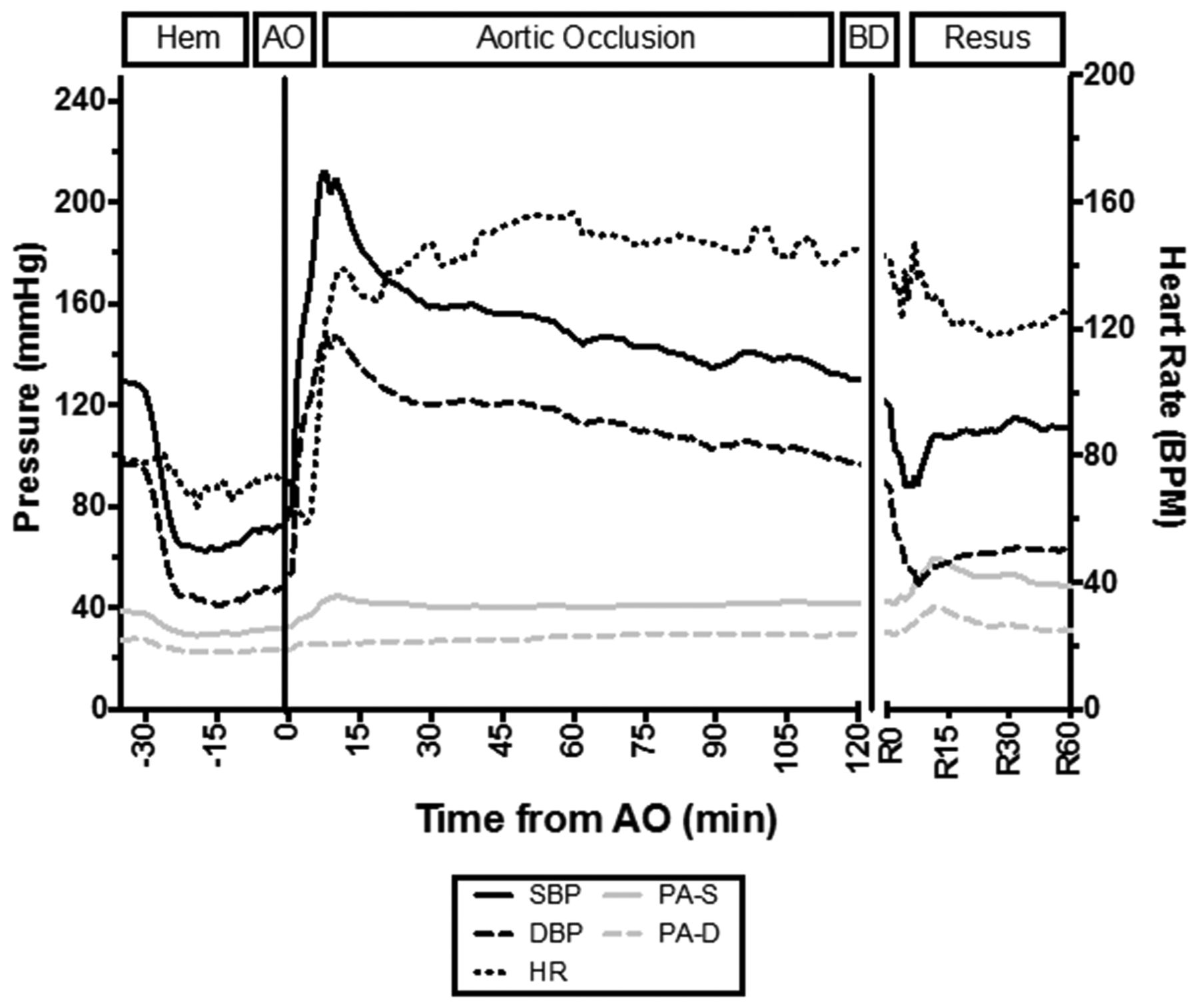
Hemodynamic responses. AO, aortic occlusion; BD, balloon deflation; bpm, beats per minute; DBP, diastolic blood pressure; hem, hemorrhage protocol phase; HR, heart rate; resus, resuscitation phase; PA-S, systolic pulmonary artery pressure; PA-D, diastolic pulmonary artery pressure; SBP, systolic blood pressure.
The immediate response to hemorrhage by the heart rate did not result in tachycardia. On balloon inflation, the swine exhibited a paradoxical episode of bradycardia for approximately 5 minutes after AO, and then became tachycardic. After balloon deflation the heart rate decreased but the animals remained tachycardic.
The arterial wave form morphology during the different phases of the experiment can be seen in figure 3A–D. After the initiation of hemorrhage, the augmentation pressure and reflective waves decrease in intensity. On balloon occlusion there is a dramatic increase in the augmentation pressure and reflective wave. In the resuscitation phase, after balloon occlusion, the reflective wave and augmentation pressure are attenuated. The AIx@75 was calculated and can be seen in figure 3E. The AIx@75 demonstrates that there is a significant and sustained supranormal increase in work and myocardial strain during REBOA. Using a repeated-measures ANOVA, the mean±SD AIx@75 during AO (T0 through T240) was significantly higher when compared with baseline (T −30; 0.44±0.10 vs. 0.27±0.05; p=0.039), hemorrhage (T-29 through T-10; 0.44±0.10 vs. 0.09±0.11; p<0.001) and resuscitation (R0 through R60; 0.44±0.10 vs. 0.20±0.16; p=0.001) phases.

Illustration of representative arterial wave forms at differing experimental time points (A–D): baseline, hemorrhage, aortic occlusion, and resuscitation, respectively. Adjusted augmentation index (AIx@75) throughout the experiment (E). AO, aortic occlusion; BD, balloon deflation; hem, hemorrhage; resus, resuscitation phase.
Myocardial injury
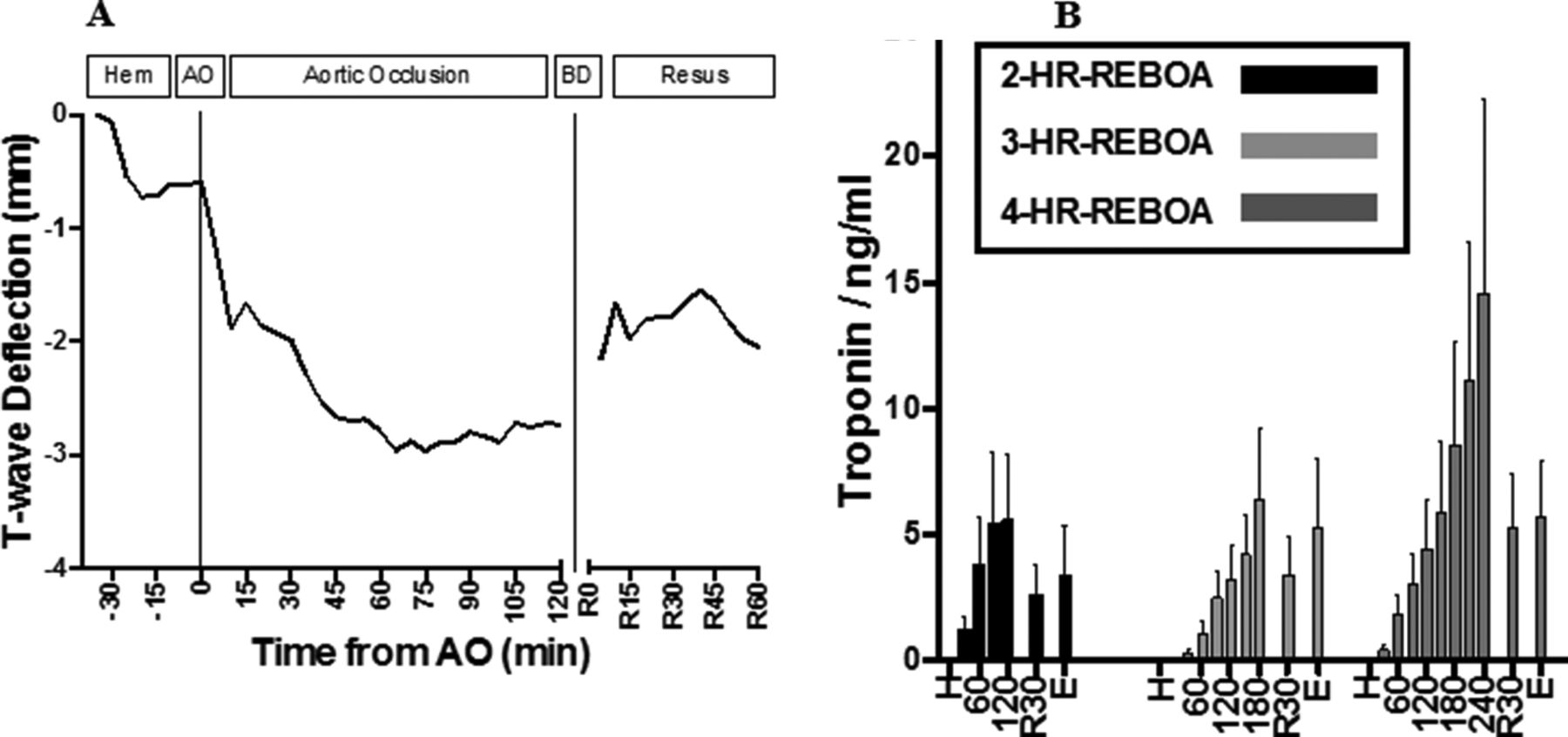
ECG tracings revealed increasing amplitude of T-wave deflection during AO that started to attenuate after balloon occlusion (see figure 4A). T-wave deflection occurred in a dose-dependent manner with respect to duration of AO (R2=0.64; p<0.001). ST-elevation or depression was not observed in the 14 studied animals. Troponin levels increased in a dose-dependent manner with respect to duration of AO (R2=0.95; p<0.001), and then started to downtrend during the resuscitation phase (see figure 4B). Histologic analysis revealed an increasing degree of inflammation and cellular injury with increasing durations of REBOA, especially in the subendocardium (see figure 5).
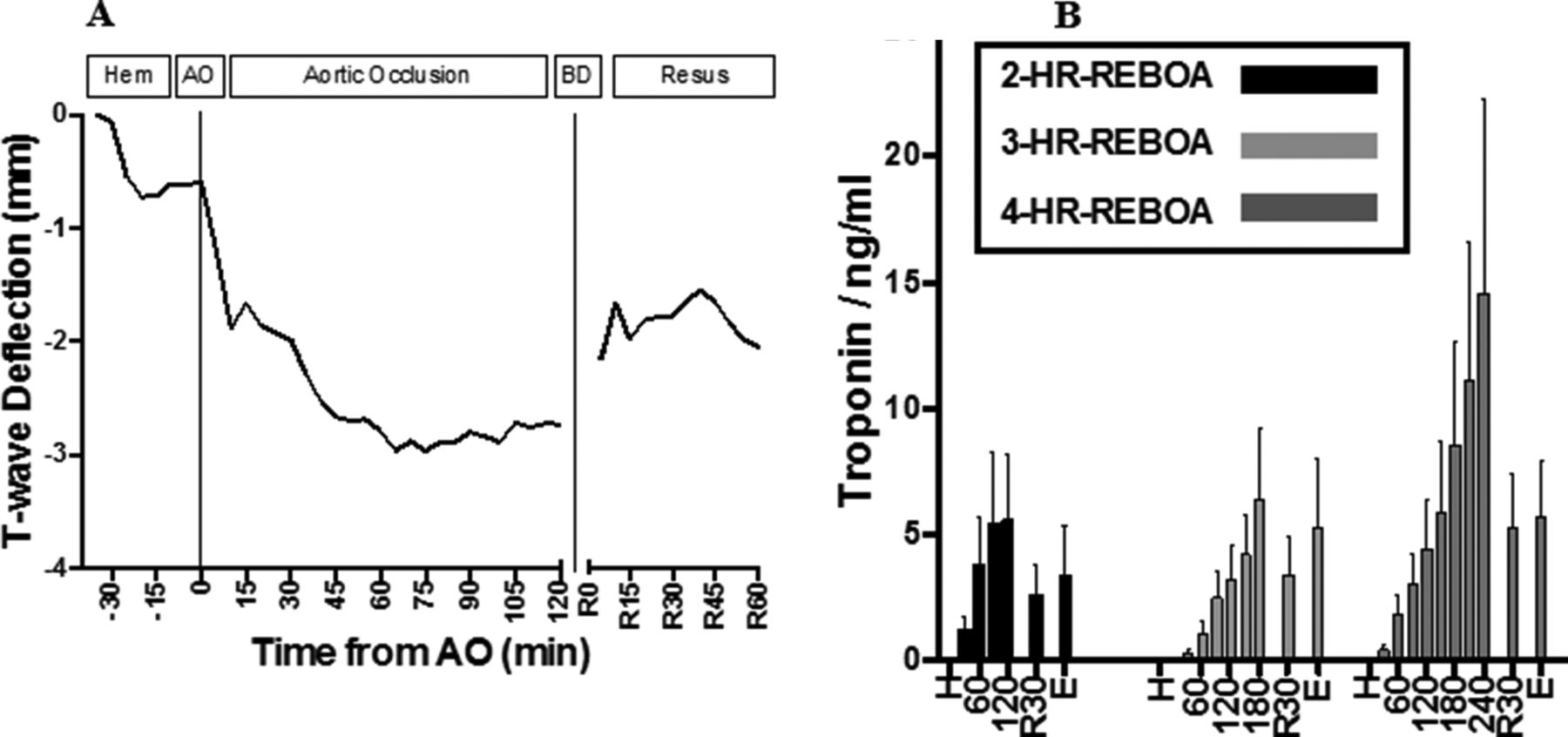
Demonstration of aggregate ECG T-wave deflection amplitude throughout the experiment (A). AO, aortic occlusion; BD, balloon deflation; hem, hemorrhage protocol phase; resus, resuscitation phase. Troponin trends for each group (B). H, hemorrhage; 2-HR-REBOA, 2 hours of occlusion; 3-HR-REBOA, 3 hours of occlusion; 4-HR-REBOA; 4 hours of occlusion; 60, 60 minutes after AO; 120, 120 minutes after AO; 180, 180 minutes after AO; 240, 240 minutes after AO; R30, 30 minutes after balloon deflation; E, 60 minutes after balloon deflation/euthanasia; REBOA, resuscitative endovascular balloon occlusion of the aorta.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Histologic incidence of myocardial eosinophilia (A). HE staining of myocardium showing subendocardial bands of ischemia (white circles, (B)). 2-HR-REBOA, 2 hours of occlusion; 3-HR-REBOA, 3 hours of occlusion; 4-HR-REBOA, 4 hours of occlusion; REBOA, resuscitative endovascular balloon occlusion of the aorta.
Discussion
This study demonstrates that REBOA, even in the setting of hypovolemia from class 4 hemorrhagic shock, results in a dramatic increase in blood pressure and afterload, which results in a duration-dependent myocardial injury. This effect is caused by REBOA through the introduction of a more proximal arterial pressure wave reflection site that effectively excludes a majority of the distal arterial tree. This leads to a sustained supranormal increase in myocardial work and strain, leading to a type 2 myocardial ischemic insult. These findings have important implications for the use of REBOA, especially in situations that require prolonged occlusion.
The mechanisms that result in myocardial ischemia secondary to severe afterload have been previously explored. Despite an increase in flow proximal to AO,6 17 an excessive increase in afterload through an amplitude increase in reflective waves (increased augmentation pressure) causes an increased workload on the heart and unfavorably alters myocardial perfusion patterns.17 18 In the context of increased afterload, coronary perfusion decreases and the patterns of flow during systole and diastole also are negatively impacted.27 28 This is caused by the early and amplified arrival of the reflected pressure wave, which in turn causes flow in late systole and diastole to diminish.17 27 In addition, these changes may lead to increased subepicardial blood flow and shunting, with reduced subendocardial blood flow,29 ultimately leading to ischemia/injury.
The AIx@75 (see figure 3E) demonstrates that there is a significant and sustained increase in work and myocardial strain during REBOA leading to myocardial injury. To provide context, the AIx@75 during AO after the initial 20 minutes in swine was higher than the AIx@75 reported for patients with unstable angina20 (46.0±9.5 vs. 38.5±8.5; p=0.04). Although an imperfect comparison, it highlights the severe penalty REBOA can impose on the heart.
These data agree with prior reports12–14 on the effects of abrupt significant increases in afterload on the heart. The results reported in our experiments likely represent the best case scenario, a non-diseased animal with a healthy heart and compliant aorta who experiences REBOA with dramatically reduced preload, which likely attenuates the afterload response from REBOA. Therefore, we hypothesize that performance of REBOA in suboptimal scenarios, such as patients with cardiovascular disease, will likely result in an even greater cardiac insult. This is supported by a study in patients undergoing infrarenal AO in which patients who had severe coronary artery disease displayed evidence of myocardial ischemia during cross-clamping, whereas those without evidence of heart disease did not.12 Additional investigations should identify ways to ameliorate the increased afterload response and effects on the heart, such as partial REBOA, as well as establish patient selection and AO duration criteria.
Although not addressed by this study, the effects of REBOA on the brain are not well characterized and may be similarly negatively impacted. Although increases in proximal flow and pressure from REBOA may exacerbate intracranial hemorrhage,30 the change in arterial impedance and capacitance may also adversely affect a non-injured brain. Similar to the effects on the heart, cases of hypertensive emergency with abrupt significant increases in afterload have been reported to result in encephalopathy, transient ischemic attack, and stroke.31 Unsurprisingly, the degree of aortic stiffness has been negatively correlated to cognitive impairment and stroke.32 33
The vasculature of the brain, which is different from most organs with respect to its perfusion patterns, provides relatively little resistance and is therefore perfused throughout systole and diastole by pulsatile flow.34 With increases in blood pressure, the cerebral arteries remain dilated and unable to dampen the increased pressures, which lead to higher wall stress, eventually leading to damage to the cerebral vasculature.34 To our knowledge, only one animal study35 has attempted to characterize the effects of REBOA on the brain. They demonstrated increased cerebral flow and pressure, but did not find that REBOA exacerbated traumatic brain injuries, although their endpoints were limited (CT imaging with very limited immediate follow-up).35 Additional research should also investigate the hemodynamic consequences of (prolonged) REBOA on the brain.
Although the current study is the first to examine myocardial injury in REBOA, there are several limitations which are important to discuss. The current study reports markers of cardiac injury, rather than cardiac function such as ejection fraction, contractility, or cardiac output. It is likely that the most important changes in functional performance would occur many hours after balloon deflation, and as this study was designed as a pilot, with a relatively short post-REBOA phase, the markers selected were thought to be most appropriate. Furthermore, the current study uses a relatively small sample size with no negative controls. The lack of control groups, specifically a group that underwent REBOA without hemorrhage and a group that underwent hemorrhage without REBOA, means that the observed cardiac injury may not be exclusively REBOA-related. Although this is possible, in the context of the literature, it is unlikely, but will require further study to be certain.
Despite these issues, the current study demonstrates that the cardiac implications of REBOA are potentially underappreciated. Future experiments should endeavor to address these limitations by using a longer follow-up period with more animals, and incorporate some functional cardiac measures with a negative control group.
Conclusions
In a swine model of hemorrhagic shock, prolonged REBOA may result in a time-dependent type 2 myocardial ischemic injury. This has important implications for patients where prolonged REBOA may be considered beneficial, and strategies to mitigate this effect require further investigation.
Acknowledgments
We would like to thank Dr Cinthia Drachenberg, who helped with histopathologic analysis. We would also like to thank Dawn Parcell for assisting with animal care.
References
Footnotes
Contributors Study conception and design: PJW, WAT, SY, HB, SMG, PH, WBG, MRH, TMS, JM. Acquisition of data: PJW, WAT, SY, SMG, PH, WBG. Analysis and interpretation of data: PJW, SY, HB, SMG, PH, JM. Drafting of the article: PJW, JM. Critical revision: PJW, WAT, SY, HB, SMG, PH, WBG, MRH, TMS, JM.
Funding This study was funded in part by a grant from the Department of Defense (grant number W81XWH-16-1-0116).
Competing interests JM is a clinical advisory board member for Prytime Medical.
Patient consent for publication Not required.
Ethics approval This study was compliant with the ethical and humane treatment of animals in research and received approval from the Institutional Animal Care and Use Committee (IACUC) at the University of Maryland, Baltimore.
Provenance and peer review Commissioned; internally peer reviewed.